In-silico follow-up results
June 29, 2020
Here we post the results from the in-silico follow up analysis done of summary statistics from release 3.
Heritability, genetic correlations and MAGMA
Summary: We created a map of candidate genomic regions associated with COVID19 phenotypes and explored the functional implications of the candidate variants/genes based on known bioinformatics sources using the functional annotation platform FUMA. We identified 12 genes associated with COVID19 phenotypes (ANKRD32, CDRT4, PSMD13, ERO1L, LZTFL1, XCR1, FYCO1, IFNAR2, CXCR6, CCR9, AP000295.9, AK5) by aggregating the variant-level association statistics into a gene-based association test using MAGMA. Based on a lookup of previous GWAS results in the gwasATLAS database, these genes are primarily implicated in immunological phenotypes. We estimated the SNP heritability of these phenotypes (0.5% - 6.6%, n.s.) using LD score regression and partitioned this heritability across functional regions of the genome, but found no significant evidence of enrichment in any functional categories. We also examined genetic correlations with an array of physiological and psychological phenotypes but found no significant genetic correlations.
Authors: Emil Uffelmann, Jeanne Savage, Philip Jansen, Christiaan de Leeuw, Danielle Posthuma
Data: CTG_insilico_COVID19-hg_29062020freeze_provisional.zip
TWAS
Summary: We ran the FUSION pipeline with the ANA_B2_V2_20200629 GWAS results. We ran FUSION using reference LD from 489 EUR 1000G individuals to perform a TWAS using total mRNA abundance in GTEx v8 Lung and WholeBlood tissues fitted with the SuSiE model. We performed a proteome-wide association study (PWAS) using SuSiE models fitted in INTERVAL plasma proteins. We also performed splice-TWAS using splice variation from GTEx v8 Lung fitted with SuSiE models.
Overall, we found two gene associations in GTEx v8 Lung:
CXCR6 (ENSG00000172215.5) twas_p=0.00000378
in total mRNA abundance GTEx v8 Lung (Bonf-p = 4.187e-06), and
IFNAR2 (ENSG00000159110.19;33252830:33262561:clu_33271) twas_p=0.00000137 IFNAR2 (ENSG00000159110.19;33230216:33244951:clu_33269) twas_p=0.0000013 IFNAR2 (ENSG00000159110.19;33246482:33246718:clu_33270) twas_p=0.00000204
in leafcutter splice variation GTEx v8 Lung (Bonf-p = 2.10e-06).
Authors: Nicholas Mancuso, Zeyun Lu, Shyamalika Gopalan, Kangcheng Hou, Bogdan Pasaniuc
Data: covid19hgi.fusion.tar.bz2
README: covid19_063020.docx
Fine Mapping
Summary: We conducted statistical fine-mapping of the meta-analysis results, assuming a single causal variant per locus and a shared causal effect across studies. For each locus with P < 1e-10 (3 Mb window around the lead variant), we applied approximate Bayes factor (ABF) with a prior variance W = 0.04 (Wakefield, J. 2009) to estimate posterior inclusion probability (PIP) and 95/99% credible sets. We observed the variants in CS at the 3p21.31 signal are in very tight LD (r2 > 0.9) across multiple populations, suggesting it might be challenging to disentangle them apart statistically. Although ABF estimates PIP = 0.85 for the lead variant of ANA_C2_V2, we note that the results should be interpreted cautiously given potential biases from different phenotyping/genotyping/imputation across studies.
COVID19_HGI_ANA_B2_V2_20200629.chr3.44367532-47367532.ABF
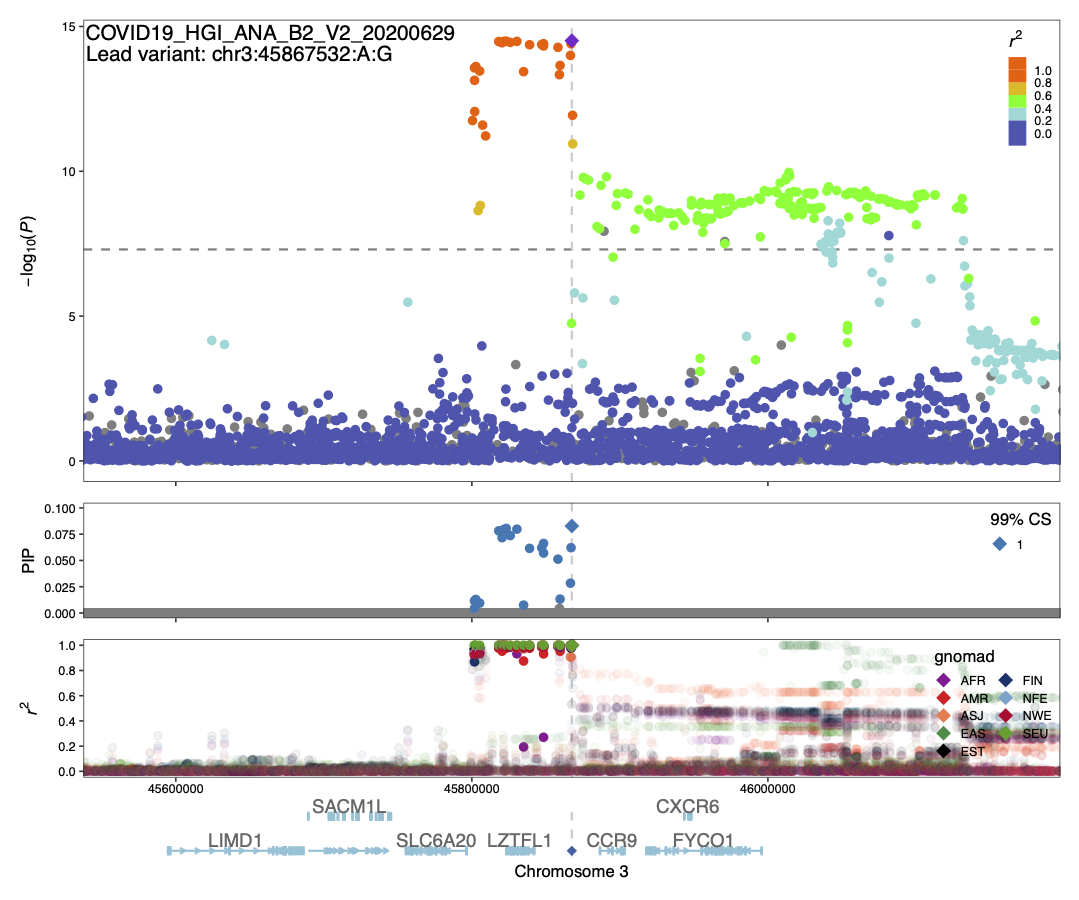
COVID19_HGI_ANA_C2_V2_20200629.chr3.44367022-47367022.ABF
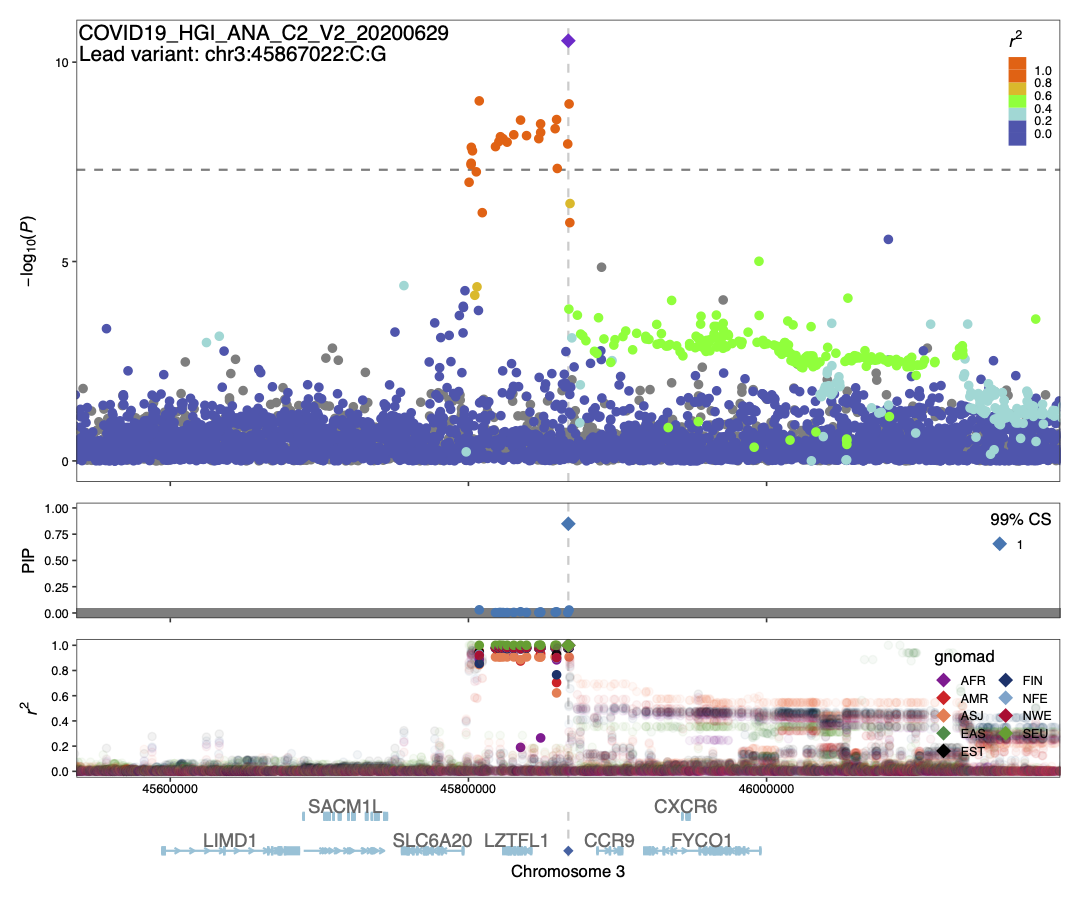
Authors: Masahiro Kanai, Hilary Finucane
Data: COVID19_HGI_20200629_ABF.tar.gz
README: README.md
Summary-data-based Mendelian Randomization
Summary: Summary-data based Mendelian Randomization (SMR) analyses of the COVID-19 GWAS summary statistics (ANA_C2_V2).
This analysis aims at detecting colocalisation of GWAS signal (p<1e-5) with methylation and expression quantitative traits loci (mQTL and eQTL respectly) from 11 mQTL and eQTL datasets (ref:https://cnsgenomics.com/software/smr/#DataResource). Top SMR results (p<1e-5) are reported. This archive contains two README files (*.txt and *.docx format), 2 SMR plots and a result file. Columns of the result file are described here:https://cnsgenomics.com/software/smr/#SMR&HEIDIanalysis.
CCR3
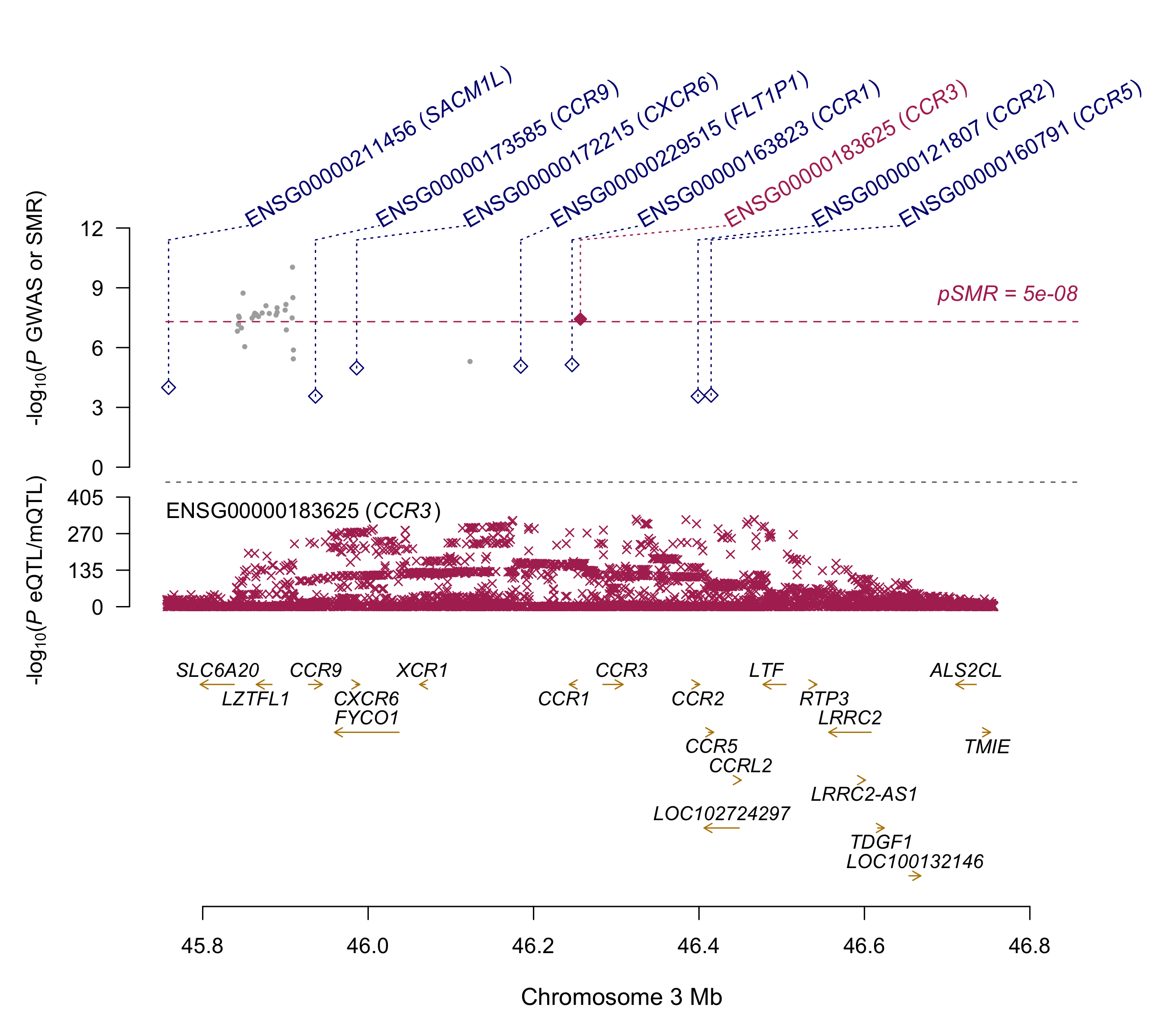
LZTFL1
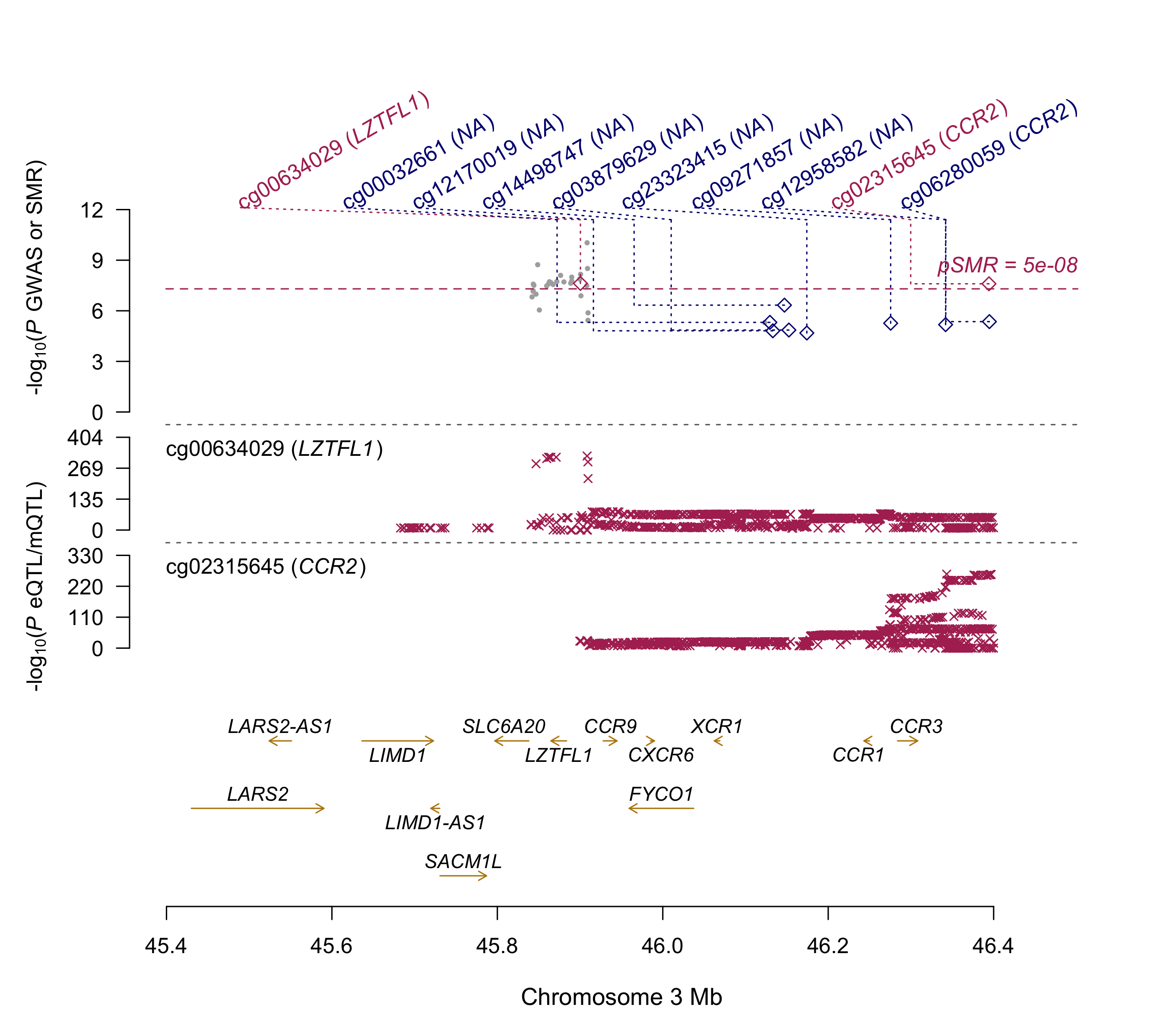
Authors: Loic Yengo, Peter Visscher & Naomi Wray
Data: ANA_C2_V2_SMR_pSMR_below_1e-5_30JUN2020_LY.txt.gz
README.docx: README.docx
README.txt: README.docx
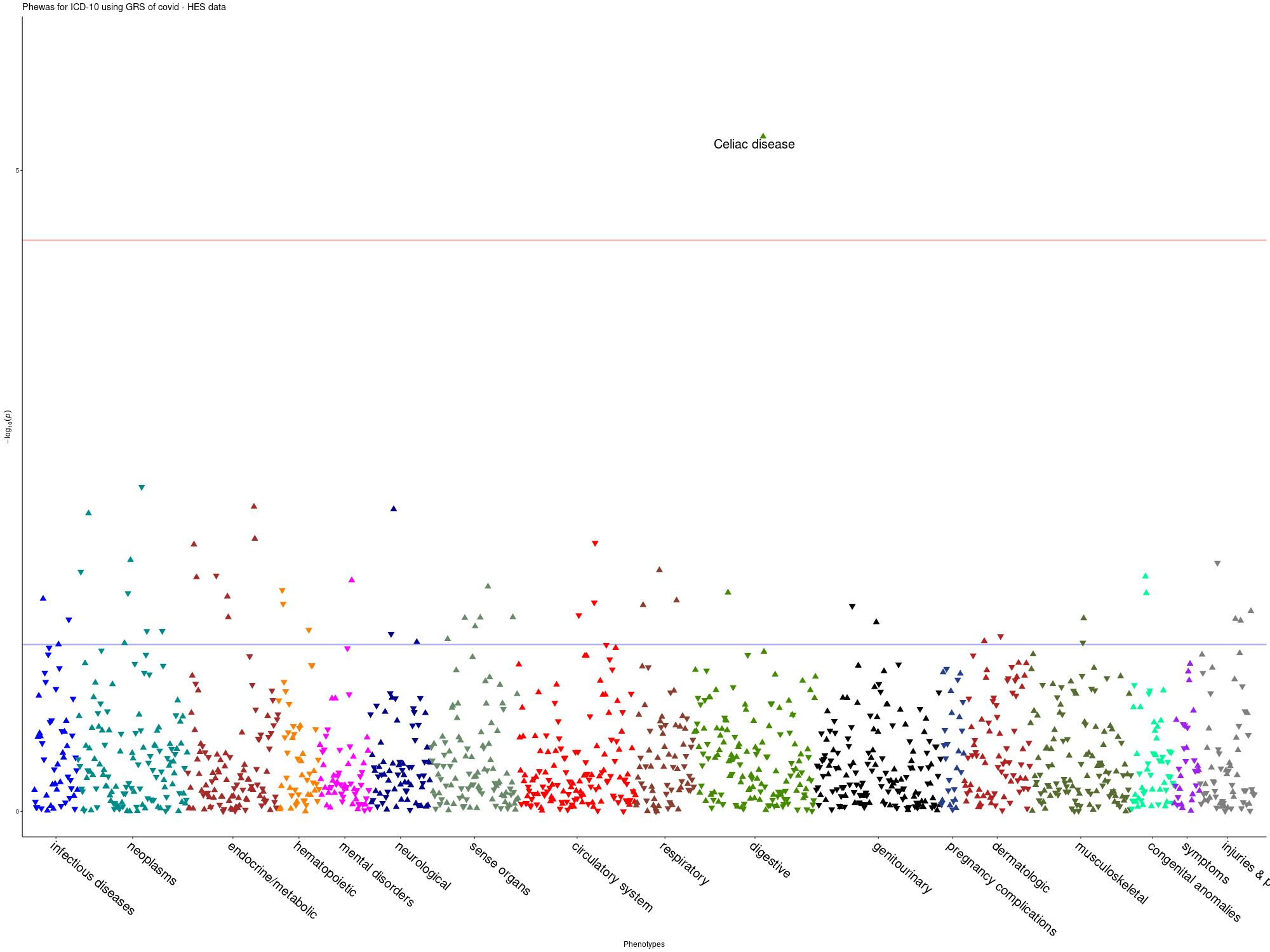
PheWAS
Summary: We performed PheWAS analyses in the UK Biobank using GP and Hospital Episode Statistics (HES) data. We used a genetic score approach based on COVID-19 associated SNPs.
ANA_B2__V2
-Genetic Score based on GWAS signals (p<1e-5). Significant outcome: Celiac Disease (p= 4.95E-07). Using summary statistics without UK Biobank, the significant outcome was Celiac Disease (p= 8.93E-07) -LD clumping was performed at SNPs with Pvalue<10-5, r2=0.1. No outcome passed Bonferroni correction.
ANA_C2__V2
-Genetic Score based on GWAS signals (p<1e-5). Marginally passing Bonferroni correction: Respiratory Arrest, P= 9.57E-05). Using summary statistics without UK Biobank, no outcome passed Bonferroni correction. -LD clumping was performed at SNPs with Pvalue<10-5, r2=0.1. No outcome passed Bonferroni correction.
ANA_C1__V2
-Genetic Score based on GWAS signals (p<1e-5). No outcome passed Bonferroni correction
Authors: Areti Papadopoulou, Eirini Marouli
Data: PheWAS_RELEASE3_2July20_EM.7z
README: PheWAS_READEME_R3_EM.txt
S-PrediXcan/MetaXcan
Summary: Variation in the human genome is expected to determine part of the individual susceptibility to COVID-19 severity and outcomes. The differences in the frequency of the genetic variants with respect to the phenotype-of-interest have small yet cumulative effect on the human transcriptome. In a transcriptome-wide association study (TWAS), the effects of genetic variants are aggregated for gene expression and tested for association with the phenotype-of-interest, reducing the multiple-testing burden with respect to a single-variant association analysis. Here, we performed TWAS using S-PrediXcan with GTEx v8 MASHR (multivariate adaptive shrinkage in R) model for lung and whole blood tissue. The MASHR models are provided at the developers host site (https://github.com/hakyimlab/MetaXcan/blob/master/README.md). The GTEx contains genotype-gene-expression data for 54 tissues from 838 donors (https://www.gtexportal.org/home/).
We performed TWAS based on the genome-wide association data for each of the six phenotypes released for COVID19-HGI Freeze3 on June 28, 2020 (https://www.covid19hg.org/results/r3/). The S-PrediXcan (alternatively known as MetaXcan) framework consists of two prediction models for GTEx v8; we used the MASHR-based model for deriving eQTL values. The MASHR model is biologically informed, with fine mapped variables (1). The variants and LD was used for EUR ancestry. The Bonferroni-corrected significance threshold accounting for the number of phenotypes, genes, and tissues tested is p-value = 2.77e-7. Significant associations were observed for BET1L (whole blood; phenotype-B1_V2 p-value = 2.33e-7) and CCR9 (whole blood; phenotype-B2_V2 p-value = 1.00e-14 | phenotype-C2_V2 p-value=9.10e-9). The detailed report is available in the link below
Authors: Gita A Pathak, Frank R Wendt, Renato Polimanti
Data
COVID-Freeze3-SMultiXcan-PathakG.xlsx
COVID-freeze3_MetaXcan_PathakG.xlsx
README