This is a translation in German. You can also read the original English version.
German-3rd-blog
April 07, 2021
COVID-19 HGI-Ergebnisse, Version 5 (Januar 2021)
- March, 2021
Geschrieben von Minttu Marttila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford, und Kumar Veerapen, ins Deutsche übersetzt von Christina Roos, Eva Schulte, Claudia Schurmann und Alexander Teumer im Namen der COVID-19 HGI
Hinweis: Die COVID-19 Host Genetics Initiative (HGI) repräsentiert ein Konsortium von über 2000 Wissenschaftlern aus über 54 Ländern, die zusammenarbeiten, um Daten und Ideen auszutauschen, Patienten zu rekrutieren und wissenschaftliche Ergebnisse zu verbreiten. Für einen Leitfaden zu unserem Studiendesign oder den Ergebnissen vom Juli 2020 (Version 3) lesen Sie bitte unseren ersten Blog-Beitrag. Unsere Forschung baut auf den vorangegangenen Ergebnissen auf, und wir fassen unsere neuesten Erkenntnisse in Blog-Beiträgen und im Ergebnisbereich unserer Website zusammen. Wenn Ihnen ein Fachbegriff nicht geläufig sein sollte, senden Sie uns bitte eine E-Mail an hgi-faq@icda.bio — wir werden die Erklärungen hier gerne aktualisieren, um das Verständnis zu erleichtern. In den kommenden Wochen werden zusätzliche Informationen zur Erläuterung von Konzepten oder Fachtermini zur Verfügung gestellt werden. In der Zwischenzeit können Sie einen Blick auf diese Seite werfen, die Ihnen die Grundlagen der Genetik ein wenig näher bringt. Wir bitten um Ihr Verständnis, dass ein Großteil der verlinkten Fachliteratur leider lediglich in englischer Sprache zur Verfügung steht.
Der wissenschaftliche Artikel zu den Ergebnissen aus Version 5 ist auf medRXiv verfügbar.
Ein größerer und robusterer Datensatz (Version 5)
Die COVID-19 HGI hat in ihren vorangegangenen Veröffentlichungen nach und nach robuste genetische Assoziationen aufgezeigt. Sie stellt bisher die größte genomweite Assoziationsstudie (GWAS) der Geschichte sowohl in Bezug auf die Studienteilnehmer (> 2 Millionen Menschen) als auch die Anzahl der Mitarbeiter (> 2.000 Wissenschaftler) dar. Hier beschreiben wir die jüngsten Ergebnisse (Version 5) der Initiative. In der vorangegangenen Version 4 berichteten wir über die Identifizierung von humangenetischen Varianten, die mit schwerer COVID-19 Erkrankung assoziiert sind (siehe unsere Blog-Beiträge für Nicht-Wissenschaftler zu Version 3 und Version 4). Wir haben diese Varianten durch eine GWAS in über 30.000 COVID-19-Patienten (d.h. Fällen) und 1,47 Millionen Menschen aus der Allgemeinbevölkerung (d.h. Kontrollen) identifiziert. In Version 5 wurde die Stichprobengröße auf fast 50.000 COVID-19-Fälle und über 2 Millionen Kontrollen erhöht, indem wir Daten aus 47 Studien aus 19 Ländern gemeinsam ausgewertet haben (Abbildung 1). Durch die Erweiterung des Stichprobenumfangs stärken wir auch das Vertrauen in unsere Ergebnisse. Weiterhin haben wir in dieser Version versucht, mehr Menschen unterschiedlicher Herkunft in unsere Studie einzuschließen. Die Untersuchung der Genetik in Populationen mit unterschiedlicher genetischer Abstammung hilft uns, genetische Varianten, die den Schweregrad von COVID-19 beeinflussen, und ihre Auswirkungen auf der ganzen Welt besser zu verstehen. Von den 47 beitragenden Studien beinhalten 19 nicht-europäische Populationen.
{kind=link}

Abbildung 1: Liste der Partner, die zur aktuellen Analyse (Version 5) de COVID-19 HGI beigetragen haben. Von den 47 beitragenden Studien umfassten 19 nicht-europäische Populationen. Angepasst aus einer Präsentation von Andrea Ganna vom 25. Januar 2021.
Aufbau der Studie
Wie in den vorherigen Versionen untersuchen wir weiterhin drei Endpunkte (Abbildung 2): A) Eine schwere COVID-19 Erkrankung bei der die Patienten beatmet werden müssen oder an COVID-19 sterben, B) eine Krankenhauseinweisung wegen COVID-19 und C) eine Infektion mit SARS-CoV-2 an sich. Diese Analysen zielen darauf ab, genetische Merkmale zu erfassen, die sowohl mit der Anfälligkeit für als auch mit dem Schweregrad der Erkrankung assoziiert sind. Die letzte Analyse (Analyse C) zielte darauf ab, genetische Varianten zu erfassen, die mit einer erfassten Infektion mit SARS-CoV-2 assoziiert sind. Diese Analyse schloss alle Fälle ein, unabhängig vom Vorhandensein oder Schweregrad von Symptomen. Die Analysen inklusive ihrer Fall- und Kontrolldefinitionen sowie Stichprobengrößen sind in Abbildung 2 dargestellt.

Abbildung 2: Definition von Fällen und Kontrollen für die einzelnen Analysen. Beachten Sie, dass SARS-CoV-2 das Virus ist, das die COVID-19-Erkrankung verursacht. Angepasst aus einer Präsentation von Andrea Ganna vom 25. Januar 2021.
Genomische Regionen, die mit COVID-19 assoziiert sind, weisen auf eine Rolle der angeborene Immunität und der Lungenfunktion hin
Nach dem Zusammenführen der genetischen Daten, die von unseren Partnern generiert wurden, führten wir die GWAS gemäß den Definitionen in Abbildung 2 durch. In Version 4 unserer Blogbeiträge hatten wir bereits neuartige genetische Signale hervorgehoben, die mit der Anfälligkeit und dem Schweregrad von COVID-19 in 7 Chromosomenregionen assoziiert sind. Im Einklang mit dem klinischen Bild der COVID-19 Erkrankung, deuten diese Regionen auf eine Beteiligung der angeborenen Immunität und der Lungenfunktion hin. In Version 5 identifizierten wir nun 15 genomweit signifikante Regionen über alle Chromosomen hinweg: Eine Chromosomenregion war in der Analyse der schweren COVID-19 Erkrankung genomweit signifikant (Analyse A); 11 Chromosomenregionen hatten einen stärkeren Effekt in der Analyse des Schweregrades als in der Analyse, in der alle Arten von SARS-CoV-2 Infektionen zusammengefasst wurden (Analyse B); und vier dieser Chromosomenregionen sind spezifisch für die Anfälligkeit für eine SARS-CoV-Infektion jedweder Schwere (Analyse C). Abbildung 3 zeigt diese Ergebnisse in Form eines Miami-Plots (eine spiegelung-basierte Version eines Manhattan-Plots, benannt nach der Stadt Miami, da sich die Skyline von Miami auf dem Wasser spiegelt).

Abbildung 3. Miami-Plot der GWAS-Ergebnisse für COVID-19. Das obere Feld zeigt die genomweiten Assoziationsergebnisse für hospitalisierte COVID-19-Patienten vs. Kontrollen (Analyse B), das untere Feld zeigt die Ergebnisse für SARS-CoV-2-Infektionen allgemein vs. Kontrollen (Analyse C).
Relevanz der unterschiedlichen Herkunft der Studienteilnehmer
Bei vielen genetischen Studien stellt der Zugang zu Proben aus vielfältigen Populationen eine Herausforderung dar (genauere Ausführung hier). Daher war die Ausweitung unserer Studie auf nicht-europäische Populationen ein zentrales Ziel der Version 5 unserer Analysen (Abbildung 4). Diese verstärkten Bemühungen haben dazu geführt, dass wir neue genetische Faktoren identifizieren konnten, die nur in bestimmten globalen Populationen mit COVID-19 assoziiert sind (unsere vorherigen Ergebnisse werden in den Blog-Beiträgen zu den Versionen 3 und 4 diskutiert). Mit unseren Analysemethoden, können wir genetische Risikofaktoren in oder in der Nähe von einzelnen oder einer Gruppe von Genen beobachten. Bisher deuten die meisten der von uns identifizierten Gene auf ein erhöhtes Risiko in den Bereichen zellulärer Mechanismen, der Immunregulation und der Herzfunktion hin. Mit der gezielten weiteren Erforschung der identifizierten Gene kann die Beschreibung dieser Risikofaktoren zur Entwicklung von neuen Therapieansätzen beitragen.
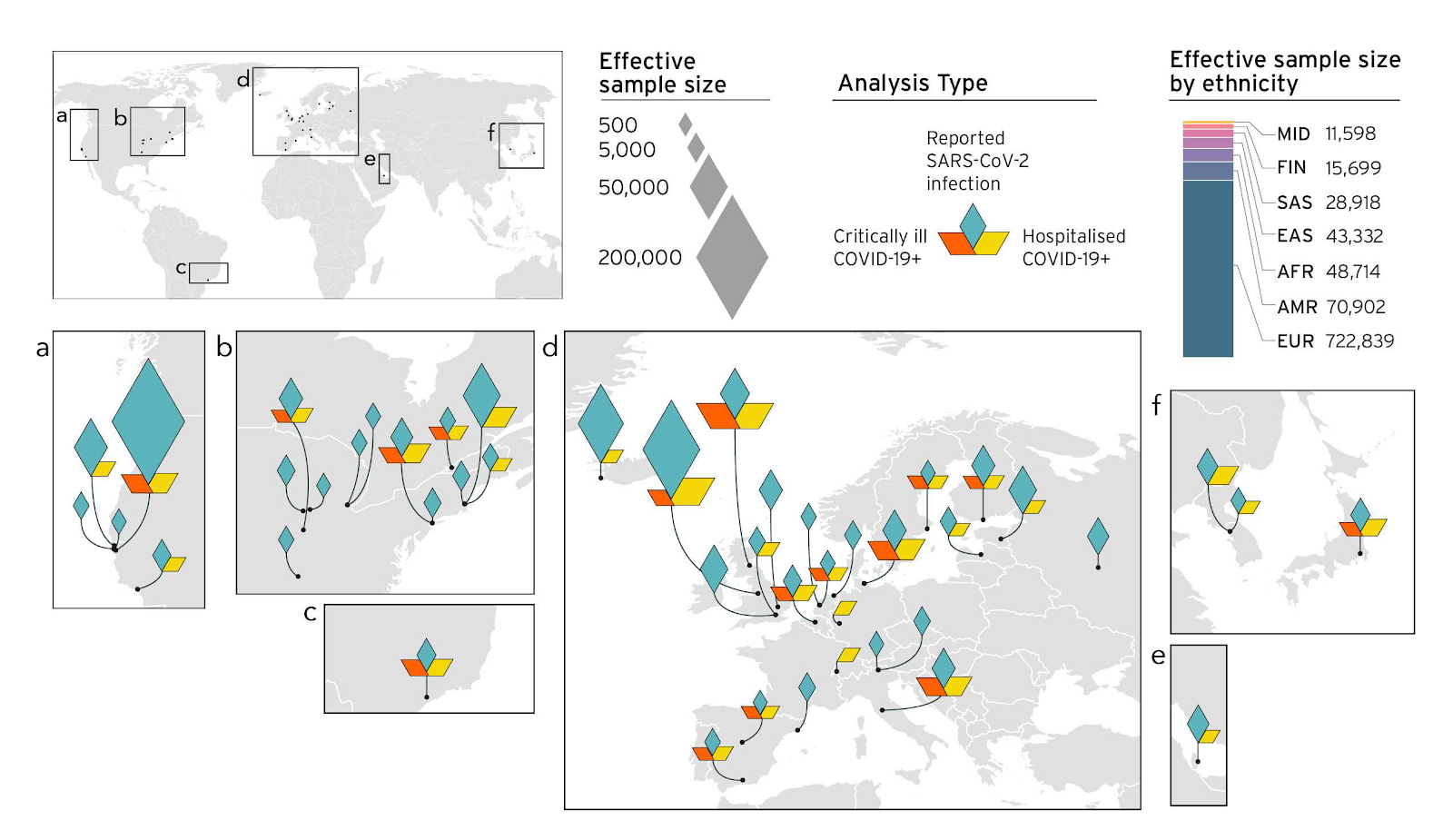
Abbildung 4. Überblick über die an der aktuellen Analyse (Version 5) der Covid-19 HGI beteiligten Studien und Zusammensetzung der Metaanalysen stratifiziert nach ethnischer Herkunft. In den aktuellen Ergebnissen sind 19 Studien mit nicht-europäischen Bevölkerungsgruppen berücksichtigt: 7 Studien trugen mit afroamerikanischen, 5 mit lateinamerikanischen, 4 mit ostasiatischen, 2 mit südasiatischen und 1 mit arabischen Populationen zur Diversität bei. Die Diamanten zeigen die Stichprobengröße an den verschiedenen Standorte weltweit.
Neun neue Chromosomenregionen, die mit COVID-19 assoziiert sind
In Analyse A (Schwer Erkrankte) sind diese in der Nähe von zwei Genen: LZTFL1 auf Chromosom 3 und TAC4 auf Chromosom 17. Das Protein LZTFL1 reguliert den Transport von Proteinen zur sogenannten Ziliarmembran. Zilien sind haarartige Strukturen, die sich außen auf der Zelle befinden. Sie kommen in den oberen Atemwegen, der Lunge und vielen anderen Organen vor. LZTFL1 ist zudem an der Immunantwort beteiligt. Das TAC4-Protein spielt eine Rolle in der Regulation von Blutdruck und Immunsystem.
In Analyse B (Patienten, mit COVID-19-bedingtem Krankenhausaufenthalt) fanden wir Assoziationen mit Varianten nahe 4 Genen: THBS3 auf Chromosom 1, SCN1A auf Chromosom 2, TMEM65 auf Chromosom 8, and KANSL1 auf Chromosom 17. Das THBS3 Gen kodiert für das Protein THBS3, das im Herzen exprimiert und bei Herzerkrankungen hochreguliert wird. Für genetische Varianten in SCN1A wurde gezeigt, dass diese mit dem Auftreten epileptischer Anfälle verbunden seinen können. TMEM65 kodiert für das TMEM65-Protein, das eine Rolle bei der Herzentwicklung, der Regulation der Reizleitung im Herzen und der Herzfunktion spielt. Möglicherweise spielt es zudem eine Rolle im zellulären Energiestoffwechsel. Interessanterweise hat die in unserer Analyse identifizierte Variante nahe/in TMEM65 eine Häufigkeit von 12% in der ostasiatischen aber nur 1% in der europäischen Bevölkerung. Die Häufigkeit unterschiedlicher Allele beschreibt das Ausmaß der Variation in einem bestimmten Gen oder einer bestimmten chromosomalen Region. Für das von KANSL1 kodierte Protein KANSL1 wird eine Beteiligung in neuronalen Prozessen vermutet.
Abschließend wurden in Analyse C (SARS-CoV-2-Infektionen jeglicher Schwere) 3 neue Assoziationen in genomischen Regionen gefunden: ZBTB11 auf Chromosom 3, DNAH5 auf Chromosom 5 und PPP1R15A auf Chromosom 19. Zunächst zeigte sich eine Assoziation mit einer Region in der Nähe des Gens ZBTB11 auf Chromosom 3. Dieses Gen kodiert für das Protein ZBTB11, von dem gezeigt wurde, dass es die Entwicklung von Immunzellen reguliert. Zweitens identifizierten wir eine chromosomale Region in DNAH5 auf Chromosom 5. Genetische Varianten in DNAH5 werden für die primäre Ziliardyskinesie, also eine seltene erbliche Erkrankung bei der es zu einer fehlerhafte Bewegung der Zilien kommt, verantwortlich gemacht. Patienten mit einer primären Ziliardyskinesie leiden an wiederkehrenden Atemwegsinfektionen, Hals-Nasen-Ohren-Symptomen und Unfruchtbarkeit. Schließlich fanden wir eine Chromosomenregion in der Nähe von PPP1R15A auf Chromosom 19. Dieses Gen kodiert für das Protein PPP1R15A, von dem gezeigt wurde, dass es Wachstumsstillstand und Zelltod als Reaktion auf DNA-Schäden, negative Wachstumssignale und falsche Proteinstrukturen vermittelt.
Gene, die das Immunsystem beeinflussen, spielen laut unserer Analysen eine wichtige Rolle bei COVID-19. Darüber hinaus und vielleicht weniger erwartet, finden sich auch Gene, die an der Lungen- und Herzfunktion sowie an neuronalen Prozessen beteiligt sind. Gut vereinbar hiermit ist, dass Herzerkrankungen bekannte Risikofaktoren für eine erhöhte Anfälligkeit für COVID-19 darstellen und neuronale Symptome als Teil der COVID-19-Krankheitsbildes beschrieben wurden.
Korrelation bedeutet nicht gleich Kausalität
Risikofaktoren, die in Beobachtungsstudien identifiziert werden, weisen möglicherweise nicht auf eine kausale Grundlage der COVID-19-Anfälligkeit oder -Schwere hin. Aus diesem Grund haben wir eine Methode namens Mendelsche Randomisierung (MR) angewendet, die genomische Informationen nutzt, um kausale Zusammenhänge abzuleiten. MR ist eine Methode, die genetische Varianten verwendet, von denen bekannt ist, dass sie ein bestimmtes Merkmal (z.B. Body-Mass-Index, BMI) beeinflussen, um den kausalen Effekt des Merkmals auf die Erkrankung zu untersuchen. Einblicke in die MR finden sich in unserem kürzlich erschienenen Blogbeitrag (für das wissenschaftliche Publikum). Über die drei COVID-19-Phänotypen hinweg identifizierten wir statistisch signifikante kausale Assoziationen zwischen den drei COVID-19-Phänotypen und 6 Merkmalen (von 38 ausgewählten und getesteten Merkmalen, Abbildung 4). Wir fanden heraus, dass ein genetisch vorbestimmter höherer BMI mit einem erhöhten Risiko für eine SARS-CoV-2-Infektion und COVID-19-Hospitalisierung einhergeht. Dieses Ergebnis bestätigt Befunde aus Beobachtungsstudien, die ein erhöhtes Risiko für schwere COVID-19-Erkrankungen in Verbindung mit einem höheren BMI beobachtet haben. Zusätzlich war auch genetisch vorhergesagtes Rauchen mit einem erhöhten Risiko für COVID-19-Hospitalisierung assoziiert.

Abbildung 5: Genetische Korrelationen und Kausalschätzer der Mendelschen Randomisierungs-Analysen zwischen 38 Merkmalen und dem COVID-19-Schweregrad bzw. einer berichteten SARS-CoV-2-Infektion. Die Merkmale sind auf der X-Achse und die COVID-19-Phänotypen auf der Y-Achse aufgeführt. Blau steht für negative genetische Korrelation und protektive kausale Effekte aus den Mendelschen Randomisierungsanalysen (MR) und rot für positive genetische Korrelation und Risiko-MR-Kausalschätzer. Größere Quadrate entsprechen einer stärkeren Korrelation. Kausalschätzer, die eine statistische Signifikanzschwelle überschreiten, sind mit einem Sternchen markiert.
Ein globales Gemeinschaftsprojekt zum besseren Verständnis der COVID-19-Wirtgenetik
In der aktuellen globalen Krise der COVID-19-Pandemie zeigen diese Ergebnisse die Stärke einer internationalen Initiative von 47 verschiedenen Partnern. Insgesamt haben wir 15 genomische Regionen identifiziert, die mit der Anfälligkeit und dem Schweregrad von COVID-19 assoziiert sind. Mit Hilfe der gefundenen genetischen Assoziationen konnten wir anhand Mendelscher Randomisierung statistisch signifikante kausale Effekte von 6 Merkmalen auf die COVID-19 Erkrankung nachweisen. Derzeit werden unsere Ergebnisse in einer wissenschaftlichen Publikation zusammengefasst. Während wir uns weiter durch die COVID-19-Pandemie kämpfen, wird die COVID-19 HGI iterativ genetische Ergebnisse produzieren. Indem wir zusammenarbeiten, können wir robuste Ergebnisse generieren, die zum besseren Verständnis der zugrunde liegenden biologischen Faktoren und dem klinischen Verlauf von COVID-19 erforderlich sind.
Danksagung
Vielen Dank an Andrea Ganna für aufmerksames Feedback und Kommentare.