This is a translation in Spanish. You can also read the original English version.
Spanish-3rd-blog
April 07, 2021
Resultados del COVID-19 HGI a enero de 2021
2 de marzo de 2021
Escrito por Minttu Marttila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford y Kumar Veerapen en nombre del COVID-19 HGI
Traducido al español por Alicia Utrilla, Carmen L Cadilla, Israel Fernández Cadenas, Macarena Fuentes, Rocío Gallego Durán, David Gómez Cabrero, Carolina Medina-Gómez, Leire Moya y Ruth Zárate.
Nota: La Iniciativa COVID-19 Host Genetics Initiative (HGI) representa un consorcio de más de 2000 científicos de 54 países que trabajan de manera colaborativa con el objetivo de compartir datos, ideas, reclutar pacientes y difundir sus hallazgos científicos conjuntamente. Si quiere conocer más sobre el diseño de nuestro estudio, por favor, lea nuestra primera publicación del blog. Nuestra investigación es interactiva y resumimos nuestros nuevos resultados a través de blog posts y en la sección de resultados de nuestra página web. Finalmente, si usted no está familiarizado con algún término aquí empleado, por favor envíenos un correo electrónico a la dirección hgi-faq@icda.bio y estaremos encantados de ampliar la información y aclarar sus dudas. En las próximas semanas, se publicará información adicional explicando conceptos y terminología usados en este documento. Mientras tanto, eche un vistazo a este recurso sobre conceptos básicos de genética.
El artículo científico sobre los resultados aquí presentados está disponible en medRXiv.
Nuestro último análisis incrementa tanto el tamaño muestral como la robustez de los hallazgos genéticos
La iniciativa COVID-19 HGI ha demostrado previamente y de manera reiterada la robusta asociación de esta enfermedad con diversas señales genéticas, y representa el mayor estudio GWAS (Genome-wide Association Study) de la historia, tanto en términos de participantes (incluye más de dos millones de individuos) como de colaboradores (participan más de 2.000 científicos). Aquí ofreceremos información de nuestro último corte de datos, el quinto en total. En cortes previos (el cuarto), se informó de la identificación de nuevas variantes genéticas en humanos asociadas con formas severas de COVID-19 (visite nuestras entradas en el blog para los no familiarizado con términos científicos (release 3 y release 4)). Hemos identificado estas variantes gracias a un estudio GWAS realizado en más de 30.000 pacientes COVID-19 (denominados casos) y en 1,47 millones de pacientes sin la enfermedad (denominados controles). En este último corte de datos, hemos incrementado el tamaño muestral a 50.000 casos y alrededor de dos millones de controles combinando datos de 47 estudios pertenecientes a 19 países (Figura 1). Al incrementar el tamaño muestral estamos mejorando la fiabilidad de nuestros resultados. Además, hemos intentado realizar también una aproximación que mejore la diversidad de las poblaciones incluidas en el estudio, ya que la evaluación genética en poblaciones de ancestros genéticos diversos incrementará el conocimiento acerca del impacto de las variantes genéticas que influyen en la severidad de la COVID-19 y su impacto a nivel mundial. De los 47 estudios incluidos, 19 de ellos incluyen poblaciones no Europeas.
{kind=link}

Figura 1: Lista de colaboradores de HGI COVID-19 del último análisis realizado (data freeze 5). De los 47 estudios, 19 incluyeron poblaciones no europeas. Adaptado de la presentación de Andrea Ganna el 25 de enero de 2021.
Estructura del estudio
Como en anteriores análisis, seguimos examinando tres resultados (Figura 2): A) Enfermos graves de COVID-19 (con asistencia respiratoria o fallecidos por COVID-19), B) Hospitalizados por COVID-19 y C) Infectados por SARS-CoV-2. Estos análisis tienen como objetivo la identificación de las características genéticas asociadas tanto a la susceptibilidad como a la severidad del SARS-CoV-2 y la COVID-19. El análisis previo (Análisis C) tuvo como objetivo detectar variantes genéticas que contribuyeran a la infección por SARS-CoV-2. Este análisis incluyó todos los casos, independientemente de la presencia o gravedad de los síntomas. Los resultados del análisis y las definiciones de casos y controles, así como el tamaño muestral, están representados en la Figura 2.
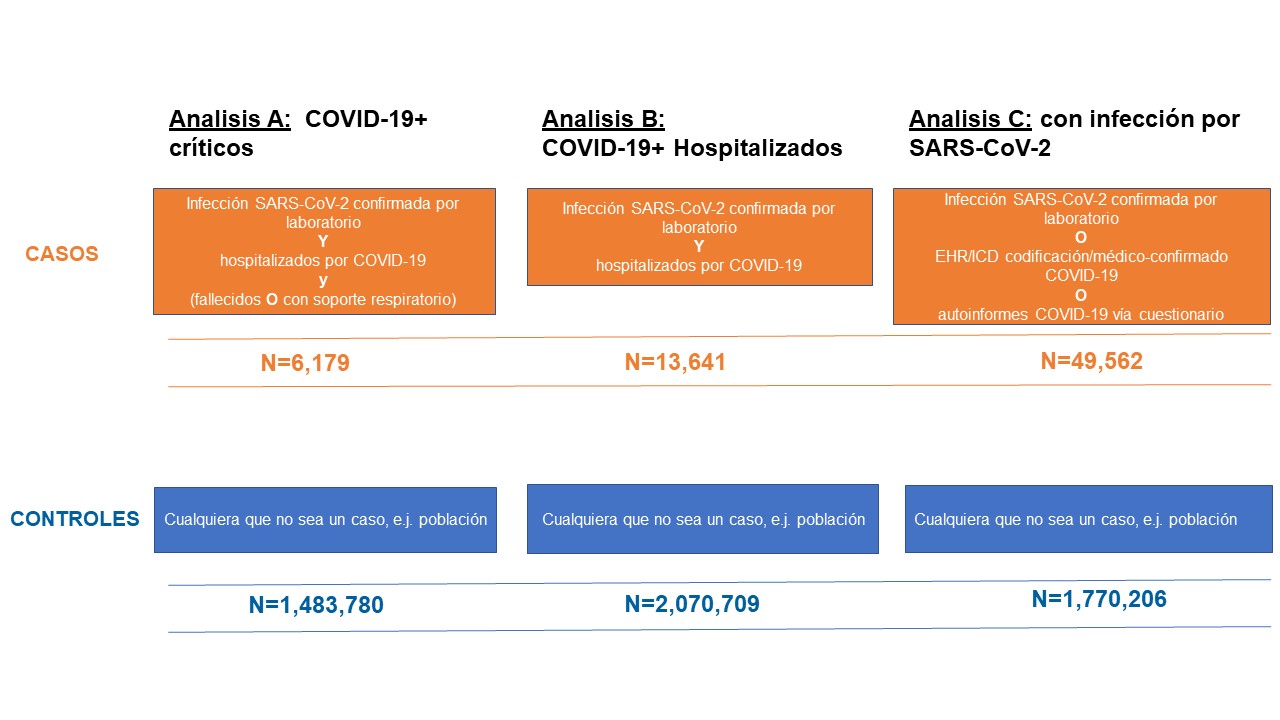
Figura 2: Definición de casos y controles para cada uno de los análisis realizados (data freeze 5). El SARS-CoV-2 es el virus que causa la infección de COVID-19. Adaptado de la presentación de Andrea Ganna el 25 de enero de 2021
Las regiones del genoma asociadas a la COVID-19 están relacionadas con la inmunidad innata y la disfunción pulmonar
Después de recopilar los datos genéticos producidos por nuestros colaboradores, realizamos un estudio GWAS de acuerdo con las definiciones de la Figura 2. En nuestros análisis previos (consulte más información en nuestra primera entrada del blog), describimos señales genéticas localizadas en 7 regiones cromosómicas asociadas a la susceptibilidad y severidad de la COVID-19. Estas regiones están relacionadas con la inmunidad innata y la disfunción pulmonar, hallazgos que van en consonancia con nuestros conocimientos clínicos actuales de la infección por COVID-19. En esta nueva versión de nuestro estudio, incluyendo más individuos (corte 5), hemos identificado de forma robusta asociaciones en 15 regiones del genoma: una de estas regiones cromosómicas fue únicamente asociada en el análisis realizado en pacientes críticamente enfermos (Análisis A); mientras que se demostró que once regiones cromosómicas tuvieron un mayor efecto sobre la severidad de la enfermedad (Análisis B) que en la susceptibilidad al desarrollo de la enfermedad (individuos que reportan haber adquirido la infección); y cuatro de estas regiones cromosómicas fueron descritas como específicas a la susceptibilidad a SARS-CoV-2 (Análisis C). En la Figura 3 se representan gráficamente estos resultados en forma de Diagrama de Miami (una versión panelizada de un diagrama de Manhattan, llamado así porque evoca cómo las edificaciones de Miami se reflejan en el agua).

Figura 3. Diagrama de Miami en el que se representan los resultados del análisis de asociación del genoma completo (GWAS) del COVID-19. El panel superior muestra los resultados del estudio entre individuos hospitalizados por COVID-19 y los controles (Análisis B), y el panel inferior los resultados del estudio entre individuos infectados con SARS-CoV-2 y los controles (Análisis C).
La importancia de la diversidad en las muestras
Entendemos que al incluir muestras de muchos estudios genéticos diferentes, la diversidad en la adquisición de las muestras es un aspecto importante a considerar (para más detalles sigue este link). Por tanto, nuestro objetivo ha sido mejorar la diversidad en la adquisición de muestras a medida que nuestro estudio crecía (Figura 4). Esta colección de muestras mejorada nos permitió identificar nuevos factores genéticos asociados a la COVID-19 (los resultados previos se analizan en las publicaciones de blog R3 y la R4). Con la identificación simultánea de factores de riesgo genéticos utilizando nuestros métodos de análisis de datos, pudimos identificar variantes genéticas dentro o cerca de regiones codificantes de genes. Hasta ahora, la mayoría de los genes que identificamos apuntan a un mayor riesgo en los mecanismos celulares, la regulación inmune y la función cardíaca. Finalmente, la identificación de estos factores de riesgo puede conducir, en última instancia, a tratamientos dirigidos a los genes identificados.
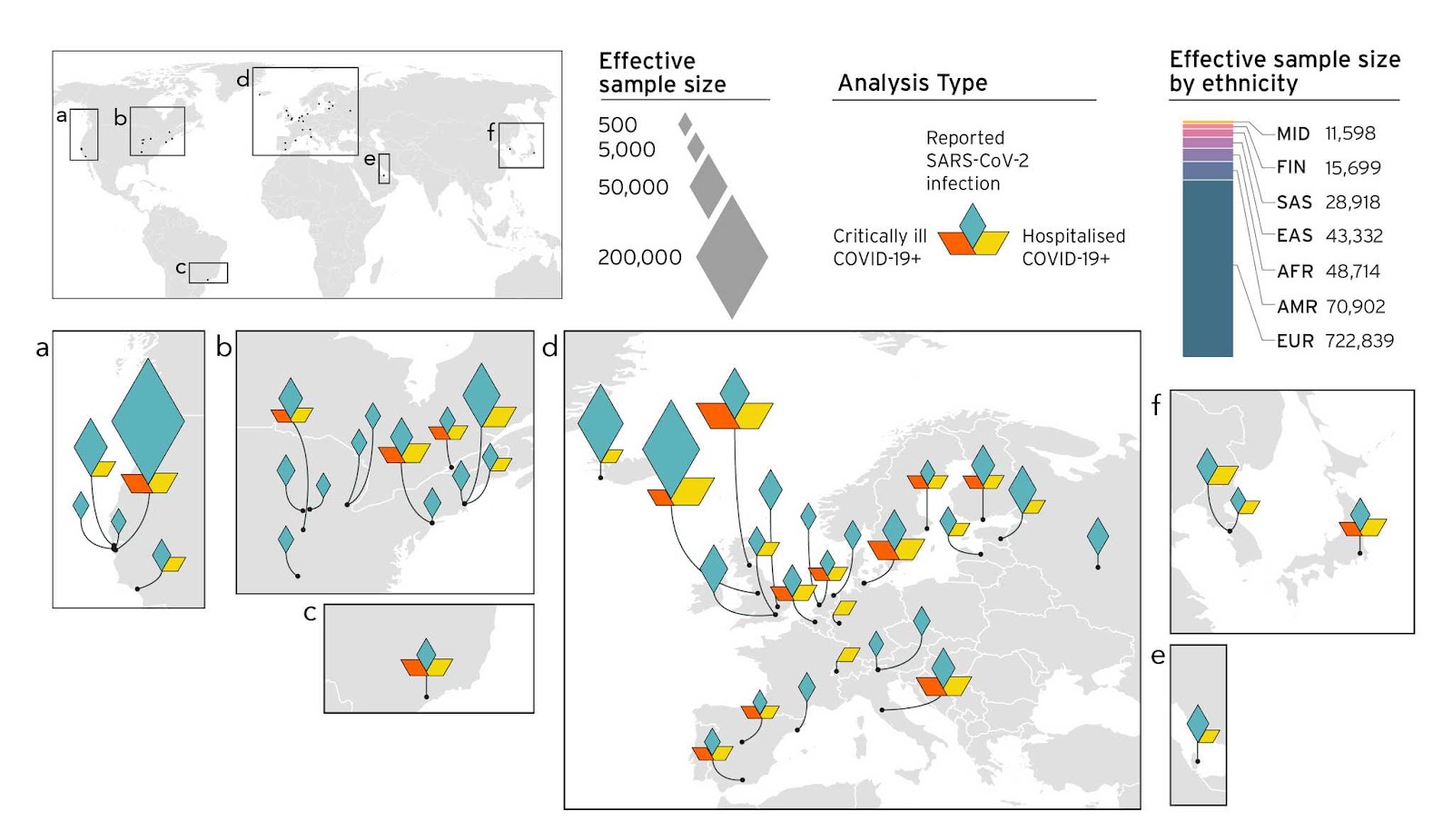
Figura 4. Perspectiva de los estudios que forman parte de la iniciativa COVID-19 HGI y composición de principales grupos de ancestros en meta-análisis. En el análisis 5, 19 estudios aportaron poblaciones no europeas: 7 de poblaciones afroamericanas, 5 de estadounidenses mixtas, 4 de Asia del Este, 2 de Asia del Sur y una población árabe. Los diamantes muestran el tamaño muestral efectivo (tamaño muestral necesario para encontrar diferencias significativas en los eventos científicos analizados) recibido de diversas localizaciones geográficas.
En este análisis, hemos encontrado 9 regiones cromosómicas nuevas asociadas con la enfermedad COVID-19. En el análisis A, realizado en enfermos críticos, se incluyeron regiones cromosómicas cercanas a dos genes: LZTFL1 en el cromosoma 3 y TAC4 localizado en el cromosoma 17. La proteína LZTFL1 regula el tráfico proteico a la membrana del cilio (desde la raíz). Los cilios son estructuras de forma similar al pelo que se encuentran en la célula. Estas proteínas se encuentran en las vías respiratorias, pulmones y otros órganos. LZTFL1 también participa en la respuesta inmune. Mientras que la proteína TAC4 tiene como funciones principales la regulación de la presión arterial y del sistema inmune.
Para el análisis B, en pacientes hospitalizados con COVID-19, encontramos asociaciones en variantes cercanas a 4 genes. En primer lugar, identificamos una región cromosómica en THBS3, localizada en el cromosoma 1. Este gen codifica la proteína THBS3 que se expresa en el corazón y aumenta su expresión en enfermedades cardíacas. En segundo lugar, identificamos una región cromosómica en el gen SCN1A en el cromosoma 2. Se ha demostrado que las variaciones en el gen SCN1A causan epilepsia y convulsiones. En tercer lugar, identificamos una región cromosómica en TMEM65 en el cromosoma 8. Este gen codifica la proteína TMEM65 que tiene un papel en el desarrollo cardíaco, y en la regulación de la conducción y función cardíacas. También puede desempeñar un papel en el metabolismo celular. Destacamos, que la variante identificada en nuestro análisis para TMEM65 tiene una frecuencia del 12% en el este de Asia y del 1% en población europea. Las frecuencias alélicas describen la cantidad de variación en un determinado gen o en una región genómica. Finalmente, identificamos una región cromosómica en KANSL1 en el cromosoma 17. Se ha sugerido que la proteína codificada por este gen, KANSL1, tiene un papel en procesos neuronales.
Finalmente, en el análisis C, el realizado en relación con la susceptibilidad a la infección por SARS-CoV-2, se han encontrado 3 nuevas asociaciones en regiones cercanas a los genes ZBTB11 en el cromosoma 3, DNAH5 en el cromosoma 5 y PPP1R15A en el cromosoma 19. En primer lugar, identificamos la región en el gen ZBTB11 en el cromosoma 3. Este gen codifica para la proteína ZBTB11,la cual regula el desarrollo de las células del sistema inmune. En segundo lugar, identificamos una región cromosómica en DNAH5 en el cromosoma 5. Se ha demostrado que las variaciones genéticas en DNAH5 causan discinesia ciliar primaria, defectos en el movimiento ciliar que conducen a infecciones torácicas recurrentes, síntomas de oído/nariz/garganta, bronquitis e infertilidad. Finalmente, hemos identificado una región cromosómica cercana al gen PPP1R15A en el cromosoma 19. Este gen codifica la proteína PPP1R15A, que se ha demostrado que media en la detención del crecimiento y la muerte celular en respuesta al daño en el ADN, señales de crecimiento negativas y estructura proteica incorrecta.
En nuestros análisis, los genes que afectan el sistema inmunológico juegan un papel importante en la COVID-19. Además, aquellos genes que afectan a la función pulmonar y cardíaca y a procesos neuronales también están incluidos en nuestros hallazgos. Las patologías cardíacas ya se habían descrito previamente como un factor de riesgo que incrementa la susceptibilidad a la COVID-19, así como a aquellos síntomas neuronales derivados de esta infección.
Correlación no significa causalidad
Cabe la posibilidad de que los factores de riesgo identificados en los estudios de asociación no apunten a las causas que generan susceptibilidad o severidad en los casos de COVID-19. Por ello, hemos empleado el método llamado Aleatorizacion Mendeliana (AM), el cual usa información genética para inferir asociaciones causales. El método AM consiste en usar variantes genéticas de las cuales se sabe que influyen en una causa concreta (por ejemplo el Índice de Masa Corporal, IMC) para examinar el efecto causal que determina el curso de una enfermedad. Para más información sobre el método de AM, lo describimos en un reciente blog post (dirigido a la audiencia científica), escrito para el público científico. De los tres fenotipos descritos para la COVID-19, hemos identificado asociaciones causales con significación estadística entre los tres fenotipos COVID-19 y seis rasgos de los 38 rasgos analizados, Figura 5). Asimismo, observamos que la predisposición genética a un IMC más elevado estaba asociada con un incremento del riesgo a ser infectado con SARS-CoV-2 y a la consecuente hospitalización por COVID-19. Este resultado confirma los hallazgos de estudios observacionales donde se ha descrito que el riesgo a presentar síntomas más severos de COVID-19 estaba asociado a un incremento del IMC. De manera similar, una predisposición genética a fumar estaba asociada con un incremento del riesgo a ser hospitalizado por COVID-19.
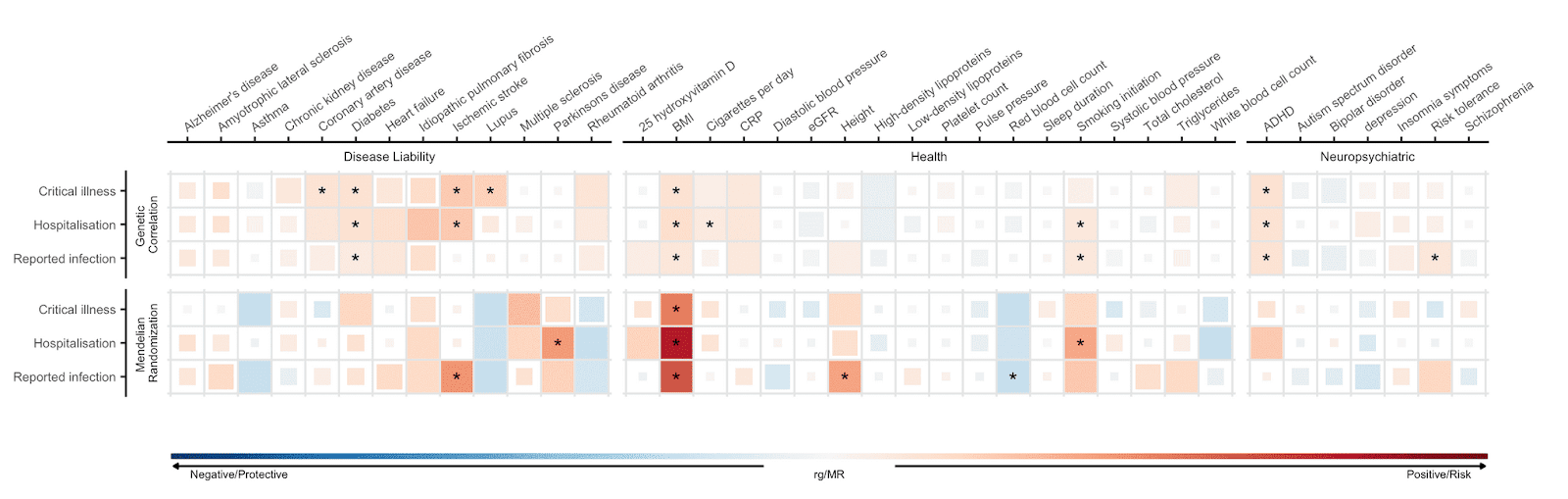
Figura 5: Correlaciones genéticas y aleatorización Mendeliana estimadas entre 38 rasgos y la severidad de la enfermedad COVID-19 y la infección por SARS-CoV-2. Estas características están representadas en el eje X, y los fenotipos COVID-19 en el eje Y. El color azul representa correlaciones genéticas negativas y aleatorización Mendeliana protectora y el color rojo representa correlación genética positiva y la aleatorización Mendeliana de riesgo. Los cuadrados de mayor tamaño corresponden a una asociación más significativa. Aquellas estimaciones causales que presentan significación estadística están marcadas con un asterisco.
Un esfuerzo colaborativo global para entender la genética del hospedador en la enfermedad COVID-19
En la crisis mundial actual debido a la pandemia del COVID-19, estos resultados demuestran un esfuerzo global de 47 estudios participantes muy diversos. En total, hemos identificado 15 regiones genómicas asociadas con la susceptibilidad y severidad de la enfermedad. Para profundizar más en la causalidad de estos hallazgos, hemos utilizado la inferencia estadística (como la aleatorización Mendeliana) para identificar 6 rasgos con causalidad estadísticamente significativa entre estas señales derivadas del GWAS realizado para COVID-19. Actualmente estamos finalizando nuestros resultados en forma de artículo científico. Mientras sigamos luchando contra esta pandemia global, la iniciativa COVID-19 HGI seguirá generando resultados genéticos de manera continuada. Al trabajar unidos podremos generar resultados sólidos que permitan mejorar el conocimiento de los factores biológicos así como de la manifestación clínica de la enfermedad COVID-19.
Agradecimientos
Gracias a Andrea Ganna, PhD, por sus comentarios y revisiones.