This is a translation in Italian. You can also read the original English version.
Italian-3rd-blog
April 07, 2021
Risultati dei dati del COVID-19 HGI (aggiornamento di Gennaio 2021)
24 Novembre 2020
Scritto da Minttu Martila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford, and Kumar Veerapen per il COVID-19 HGI
Tradotto da: Francesca Colombo, Margherita Francescatto, Pietro Lio', Kathrin Aprile von Hohenstaufen Puoti, Pasquale Striano (0000-0002-6065-1476), Constanza Vallerga
Caveat: il COVID-19 Host Genetics Initiative (HGI) rappresenta un consorzio di oltre 1000 scienziati provenienti da oltre 54 paesi che lavorano insieme per condividere dati, idee, reclutare pazienti e divulgare i risultati delle ricerche. Per un'introduzione al progetto ed ai risultati del Luglio 2020, si rimanda al post specifico (inaugural blog post). La ricerca è ultimata ed i ‘results’ sono in fase di pubblicazione (https://www.medrxiv.org/content/10.1101/2021.03.10.21252820v1.article-info). Infine, se qualche vocabolo o concetto non fosse chiaro o poco familiare, inviaci un'e-mail a hgi-faq@icda.bio e saremo lieti di aggiornare le informazioni per far chiarezza. Nelle prossime settimane saranno rese disponibili ulteriori informazioni che spiegano concetti o terminologia. Se necessario, puoi dare un'occhiata a questa risorsa (this) per rivedere le basi della genetica.
L'ultimo aggiornamento dei dati (release 5) aumenta la dimensione del campione e consolida i risultati dello studio genetico
Il consorzio COVID-19 HGI ha riportato, ripetutamente nelle precedenti release, robusti segnali di associazione genetica. Inoltre, rappresenta il più grande studio di associazione genome-wide (GWAS) della storia, sia in termini di partecipanti allo studio (>2 milioni di individui) che di numero di collaboratori (> 2.000 scienziati). Qui, descriviamo i risultati aggiornati della nostra ultima release 5. Nella precedente (release 4), avevamo riportato l'identificazione di varianti genetiche umane associate a COVID-19 grave (si vedano i nostri post sul blog per non scienziati, sulle release 3 e 4). Avevamo identificato queste varianti attraverso un GWAS in oltre 30.000 pazienti COVID-19 (cioè i casi) e 1,47 milioni di pazienti non-COVID-19 (cioè i controlli). Nella release 5, abbiamo ulteriormente aumentato la dimensione del campione a quasi 50.000 casi COVID-19 e oltre 2 milioni di controlli, combinando i dati di 47 studi in 19 paesi (Figura 1). Aumentando la dimensione del campione, abbiamo anche migliorato la confidenza dei nostri risultati. Inoltre, abbiamo anche cercato di ampliare la diversità delle popolazioni analizzate. Lo studio della genetica tra popolazioni di diverse origini genetiche ci aiuta a capire meglio le varianti genetiche che influenzano la gravità del COVID-19 e il loro impatto in tutto il mondo. Dei 47 studi che hanno contribuito, 19 includono popolazioni non europee.
{kind=link}
Figura 1: Lista dei collaboratori del consorzio COVID-19 HGI che hanno contribuito alla release 5. Dei 47 studi che hanno contribuito, 19 includono popolazioni non europee. Adattata dalla presentazione di Andrea Ganna.
Struttura dello studio
Come nei precedenti aggiornamenti, abbiamo condotto analisi per trovare fattori genetici associati a (Figura 2):
A) Essere stati gravemente malati di COVID-19 (situazione definita dalla necessità di supporto respiratorio oppure dall’esito di decesso per COVID-19),
B) Essere stati ricoverati per COVID-19
C) Essere stati infettati da SARS-CoV-2.
Queste analisi sono volte a identificare le caratteristiche genetiche associate sia alla suscettibilità all’infezione da SARS-CoV-2, che alla gravità del COVID-19. In particolare, l'ultima analisi (C) mira specificamente a rilevare le varianti genetiche che contribuiscono all'infezione da SARS-CoV-2 ed ha incluso tutti i casi, indipendentemente dalla presenza o dalla gravità dei sintomi, purché avessero avuto il COVID-19 (confermato mediante indagine di laboratorio o da un medico oppure riferito dal paziente stesso). L’oggetto delle tre analisi, le definizioni di caso e controllo e le dimensioni del campione per ciascuna di esse sono mostrati nella Figura 2.

Figura 2: Definizione dei casi e dei controlli in ciascuna delle tre analisi condotte nella release 5. (dalla presentazione di Andrea Ganna del 25 gennaio 2021).
Le regioni del genoma associate al COVID-19 implicano l’immunità innata e la disfunzione polmonare
Una volta raccolti i dati genetici generati dai nostri collaboratori, abbiamo eseguito uno studio GWAS in linea con le definizioni riportate in Figura 2. In precedenza, nella release 4 (data freeze 4), si erano evidenziati i nuovi segnali genetici associati alla suscettibilità e severità del COVID-19 in 7 regioni cromosomiche. Queste regioni puntano sull’eziologia dell'immunità innata e della disfunzione polmonare: in linea con la principale comprensione clinica delle infezioni da COVID-19. Nella release 5, abbiamo identificato 15 regioni genomiche significative tra tutti i cromosomi: una regione cromosomica è risultata significativa solo nell' analisi genomica sulla condizione di malattia grave da COVID-19 (analisi A); 11 regioni cromosomiche sono risultate avere un effetto maggiore nell' analisi genomica sulla severità da COVID-19 rispetto a quello individuato dall’analisi effettuata sulle infezioni da SARS-CoV-2 (analisi B); e 4 regioni cromosomiche sono risultate specifiche dell'infezione riportata da SARS-CoV-2 (analisi C). Nella Figura 3, è illustrata una rappresentazione grafica di questi risultati sotto forma di grafico denominato “Miami plot” (una versione del Manhattan plot suddivisa in pannelli. Il nome deriva dalla città di Miami la cui linea d’orizzonte si riflette sull'acqua).
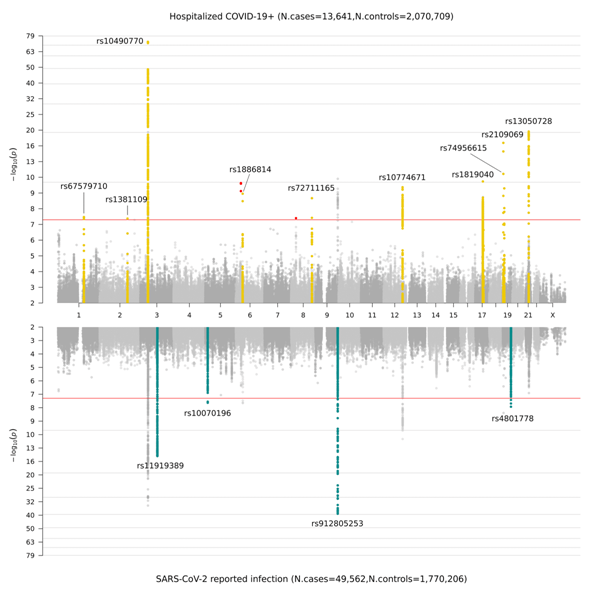
Figure 3. “Miami plot” che mostra i risultati dello studio GWAS sul COVID-19. Il pannello superiore mostra I risultati dello studio GWAS condotto su pazienti ospedalizzati per COVID-19 e controlli (analisi B); il pannello inferiore mostra i risultati dello studio GWAS condotto su pazienti con dichiarate infezioni da SARS-CoV-2 e controlli (analisi C).
Il ruolo della diversità dei campioni
In molti studi genetici la varietà nella collezione dei campioni ricopre un ruolo cruciale (maggiori dettagli sono disponibili qui). Per questo motivo, mano a mano che il nostro studio si è ampliato ci siamo sforzati per migliorare la diversità dei campioni raccolti (Figura 4). Questo ci ha permesso di identificare nuovi fattori genetici associati a COVID-19 (i risultati ottenuti in precedenza sono discussi nei relativi blog posts Release 3 e Release 4). Con la concomitante identificazione di fattori di rischio genetico utilizzando i nostri metodi analitici, abbiamo anche la capacità di individuare varianti genetiche dentro o nelle vicinanze di geni. Finora, la maggior parte dei geni che abbiamo identificato suggeriscono che l’aumento del rischio è legato a meccanismi cellulari, alla regolazione dell’attività immunitaria e della funzione cardiaca. L’individuazione di tali fattori di rischio può permettere in definitiva di sviluppare trattamenti specifici indirizzati verso i geni identificati.

Figura 4. Panoramica degli studi che contribuiscono alla COVID-19 host genetics initiative e composizione secondo i principali gruppi di ascendenza genetica nelle metanalisi. Nell’analisi data freeze 5 sono stati inclusi 19 studi con popolazioni non-Europee: 7 Afroamericana, 5 Americana mista, 4 Asia dell’Est, 2 Asia del Sud ed 1 Araba. I diamanti mostrano la dimensione effettiva del campione (dimensione del campione che identifica l’effetto statisticamente significativo negli eventi scientifici) ricevuta da diverse località geografiche.
Abbiamo identificato 9 nuove regioni cromosomiche associate a COVID-19. Nell’analisi A, per malattia critica, le associazioni includono regioni vicino a due geni: LZTFL1 sul cromosoma 3 e TAC4 sul cromosoma 17. La proteina LZTFL1 regola il movimento delle cilia, protrusioni sottili che si estendono dal corpo cellulare e particolarmente importanti nelle vie respiratorie e nei polmoni. LZTFL1 partecipa anche alla risposta immunitaria. La proteina TAC4 è invece coinvolta nella regolazione della pressione sanguigna e nel sistema immunitario.
Per l’analisi B, in pazienti ospedalizzati con diagnosi di COVID-19, abbiamo trovato associazione in varianti situate vicino a 4 geni. Per prima cosa abbiamo identificato una regione cromosomica su THBS3 sul cromosoma 1. Questo gene codifica per la proteina THBS3 che è espressa nel cuore ed è sovraespressa nelle malattie cardiache. In secondo luogo abbiamo identificato una regione cromosomica in SCN1A sul cromosoma 2. E’ stato dimostrato che varianti nel gene SCN1A causano epilessia od encefalopatia epilettica. Abbiamo anche identificato una regione cromosomica in TMEM65 sul cromosoma 8. Questo gene codifica per la proteina TMEM65 che è coinvolta nello sviluppo del cuore e nella regolazione della conduzione e della funzione cardiaca. E’ possibile che abbia anche un ruolo nel metabolismo energetico delle cellule. E’ da notare che la variante in TMEM65 identificata mediante le nostre analisi ha frequenza del 12% nell’Asia dell’Est e dell’1% nella popolazione europea. In generale la frequenza allelica descrive quanta variazione è stata osservata in una data regione genomica. Abbiamo infine identificato una regione cromosomica in KANSL1 sul cromosoma 17. Si ritiene che la proteina codificata da questo gene, KANSL1, abbia un ruolo nei processi neuronali.
Infine, nell’analisi C, per infezioni SARS-CoV-2 riportate, sono state identificate tre nuove associazioni in regioni vicino ai geni ZBTB11 sul cromosoma 3, DNAH5 sul cromosoma 5, e PPP1R15A sul cromosoma 19. In primo luogo, abbiamo identificato una regione vicino al gene ZBTB11 sul cromosoma 3. Questo gene codifica per la proteina ZBTB11 che è coinvolta nella regolazione dello sviluppo delle cellule del sistema immunitario. Abbiamo poi individuato una regione sul cromosoma 5 vicino al gene DNAH5. E’ stato dimostrato che varianti genetiche in DNAH5 portano a infezioni polmonari ricorrenti, sintomi legati a orecchie/naso/gola, bronchite e infertilità. Infine abbiamo identificato una regione cromosomica vicino al gene PPP1R15A sul cromosoma 19. Questo gene codifica per la proteina PPP1R15A, coinvolta nell’ arresto della crescita e nella morte cellulare in risposta a danni del DNA, segnali negativi di crescita e incorretta struttura proteica.
Nella nostra analisi, geni che influenzano il sistema immunitario giocano un ruolo importante. Sono parte delle nostre scoperte anche geni coinvolti nella funzionalità polmonare e cardiaca e in processi neurologici. Le malattie cardiache erano state precedentemente segnalate come fattore di rischio per COVID-19 e sintomi neurologici erano stati riportati come parte della malattia COVID-19.
‘Correlazione’ non significa necessariamente ‘causa’
I fattori di rischio identificati negli studi di associazione non possono essere posti in una sicura relazione causale di suscettibilità o severità’ per il COVID19. L’ analisi di randomizzazione mendeliana (Mendelian randomization, MR) fa uso dell’ informazione genomica per stabilire un nesso causale. Questo metodo si basa sull'influenza che le varianti genetiche hanno su un aspetto fenotipico, ad esempio l’ indice di massa corporea (BMI), per l’ esame del nesso causale di tale fattore fenotipico con lo stato della malattia. Una descrizione tecnica di questo metodo si trova in questo recente post. L'analisi dei tre stati di COVID19 ci ha portato a stabilire che associazioni causali tra questi tre stati di COVID-19 e i sei tratti fenotipici sono statisticamente robuste (rispetto ai 38 tratti fenotipici selezionati che sono stati testati; vedi Figura 4). Abbiamo trovato che la predizione su base genetica di un valore più’ alto di indice di massa corporea (BMI) e’ associata ad un maggiore rischio di infezione da SARS-CoV-2 e di ospedalizzazione per COVID-19. Questi risultati confermano precedenti studi osservazionali su un maggiore rischio di severità’ di infezione COVID-19 associato con un maggiore indice di massa corporea. Inoltre, la predizione di abitudine a fumare, fatta su base genetica e’ associata ad un maggiore rischio di ospedalizzazione per COVID-19.
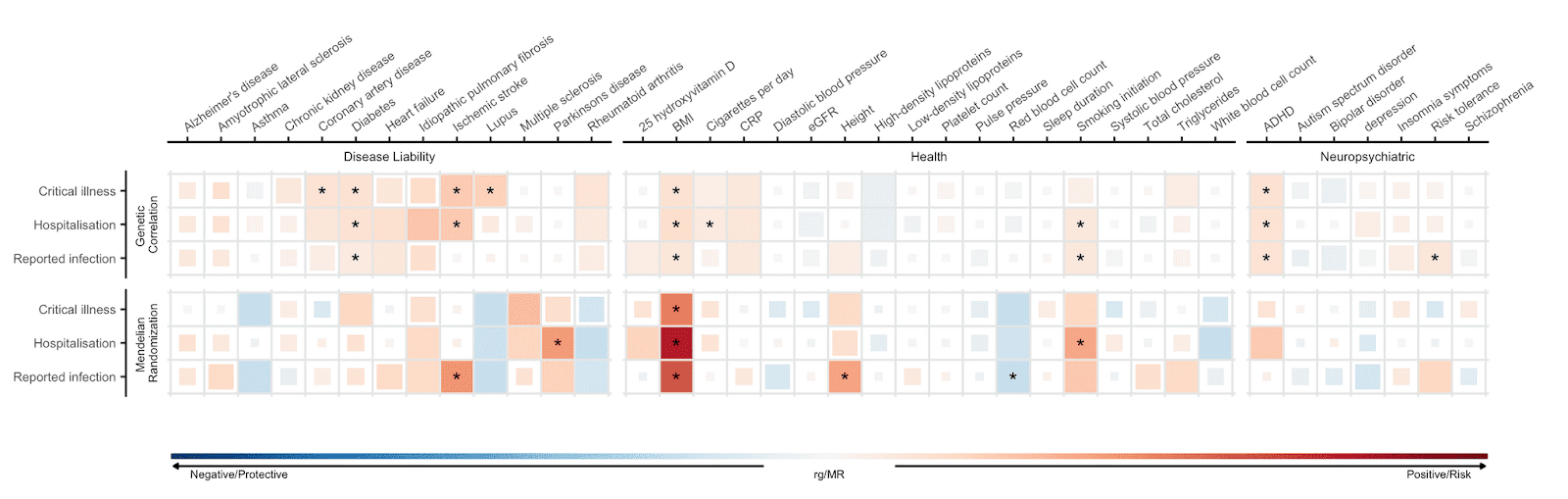
Figura 5: Correlazione genetica e la statistica di causalità basata sul metodo di randomizzazione Mendeliana tra 43 tratti fenotipici e la severità dello stato clinico di COVID-19 e dell'infezione da SARS-CoV-2. I tratti sono mostrati sull’ asse delle X e i fenotipi di COVID-19 sono sull’asse Y. Il colore blu rappresenta una correlazione genetica negativa e un effetto protettivo determinato con il metodo della randomizzazione Mendeliana; il colore rosso rappresenta una correlazione positiva ed un fattore di rischio. Quadrati grandi corrispondono ad una correlazione più forte. Gli asterischi mostrano i risultati per i quali la stima di causalità è statisticamente significativa.
Uno sforzo collaborativo globale per comprendere la genetica dei soggetti affetti da COVID-19
Questi risultati dimostrano l’efficacia di uno sforzo globale di 47 diversi gruppi di lavoro nella presente crisi globale della pandemia da COVID-19. Abbiamo identificato in tutto 15 regioni genomiche associate alla suscettibilità e severità della malattia COVID-19. Per indagare ulteriormente la relazione causale di queste regioni abbiamo utilizzato l’inferenza statistica (ossia la Mendelian randomization) identificando 8 tratti con causalita statisticamente significativa relativamente a questi segnali dell’analisi GWAS. Al momento stiamo finalizzando i nostri risultati in un articolo scientifico. Mentre proseguiamo la marcia attraverso la pandemia da COVID-19, il consorzio COVID-19 HGI continuerà a produrre ripetutamente risultati di analisi genetica. Lavorando insieme possiamo generare scoperte robuste necessarie per comprendere meglio i fattori biologici e la presentazione clinica dell’infezione COVID-19.
Ringraziamenti
Un grazie speciale ad Andrea Ganna, PhD per il coordinamento del Consorzio.