This is a translation in Dutch. You can also read the original English version.
Dutch-3rd-blog
April 07, 2021
De COVID-19 HGI resultaten vanaf januari 2021
Geschreven door Minttu Marttila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford, en Kumar Veerapen namens het COVID-19 HGI consortium.
Vertaald naar Nederlands door Philip Jansen, Sander van der Laan, Cindy Boer en Lude Franke.
Disclaimer: Het COVID-19 Host Genetics Initiative (HGI) vertegenwoordigt een consortium van meer dan 2.000 wetenschappers uit 54 landen die samenwerken om de genetica van het risico op COVID-19 te onderzoeken. Binnen COVID-19 HGI wisselen wij data en ideeën uit, verzamelen en delen wij patiënten data (met toestemming), en delen we de bevindingen van het onderzoek via (sociale) media en wetenschappelijke tijdschriften met de maatschappij. Voor een introductie over de opzet van de studie, verwijzen we naar de eerdere blog post. Dit onderzoek wordt herhaaldelijk geüpdatet: we vatten steeds nieuwe resultaten samen in blog posts en in de resultatensectie op onze website. Wanneer de betekenis van specifieke woorden, die hier gebruikt worden, niet duidelijk is, stuur ons dan gerust een e-mail via hgi-faq@icda.bio. We zullen dan zorgen dat de tekst duidelijker wordt gemaakt en geven graag extra toelichting hierop. In de komende weken zal extra informatie beschikbaar worden gemaakt over de besproken concepten en gebruikte terminologie. In de tussentijd kan deze bron worden gebruikt als toelichting op de basisbegrippen die in de genetica gebruikt worden.
Het wetenschappelijke artikel waar deze blog post op gebaseerd is, is ook online te vinden op medRXiv
Deze laatste versie (release 5) vergroot de studieomvang en bevordert de betrouwbaarheid van de bevindingen
Het onderzoek van COVID-19 HGI heeft meerdere robuuste genetische associaties opgeleverd in de vorige versie en is momenteel de grootste genoom-brede associatie studie (GWAS) in de geschiedenis, zowel qua aantallen deelnemers (>2 miljoen deelnemers) als het aantal wetenschappers dat bij het project betrokken is (>2,000). In deze blog beschrijven we de resultaten van onze laatste versie (release 5). In de vorige versie (release 4), rapporteerden wij meerdere genetische varianten die verband houden met een ernstig ziektebeloop van COVID-19 (zie onze blog posts voor niet-wetenschappers van release 3 en release 4). We identificeerden deze varianten door een GWAS uit te voeren in meer dan 30.000 COVID-19 patiënten (‘gevallen’, in het Engels ‘cases’) en 1.47 miljoen gezonde vrijwilligers die geen COVID-19 hadden (‘controles’). In deze nieuwe versie release 5, hebben we de studieomvang verder kunnen uitbreiden naar bijna 50.000 COVID-19 gevallen en meer dan 2 miljoen controles door het combineren van 47 studies die in 19 verschillende landen werden uitgevoerd (zie Figuur 1). Door de toename in het aantal deelnemers neemt de betrouwbaarheid van de resultaten toe. In deze versie hebben we ook geprobeerd meer verschillende populaties in de studie toe te voegen. Door genetische variatie in populaties van verschillende etnische achtergronden te bestuderen, kunnen we de genetische varianten die van invloed zijn op het COVID-19 beloop beter begrijpen in bevolkingen op verschillende plaatsen van de wereld. Van de 47 deelnemende studies zijn er 19 die niet-Europese populaties hebben geïncludeerd.
{kind=link}
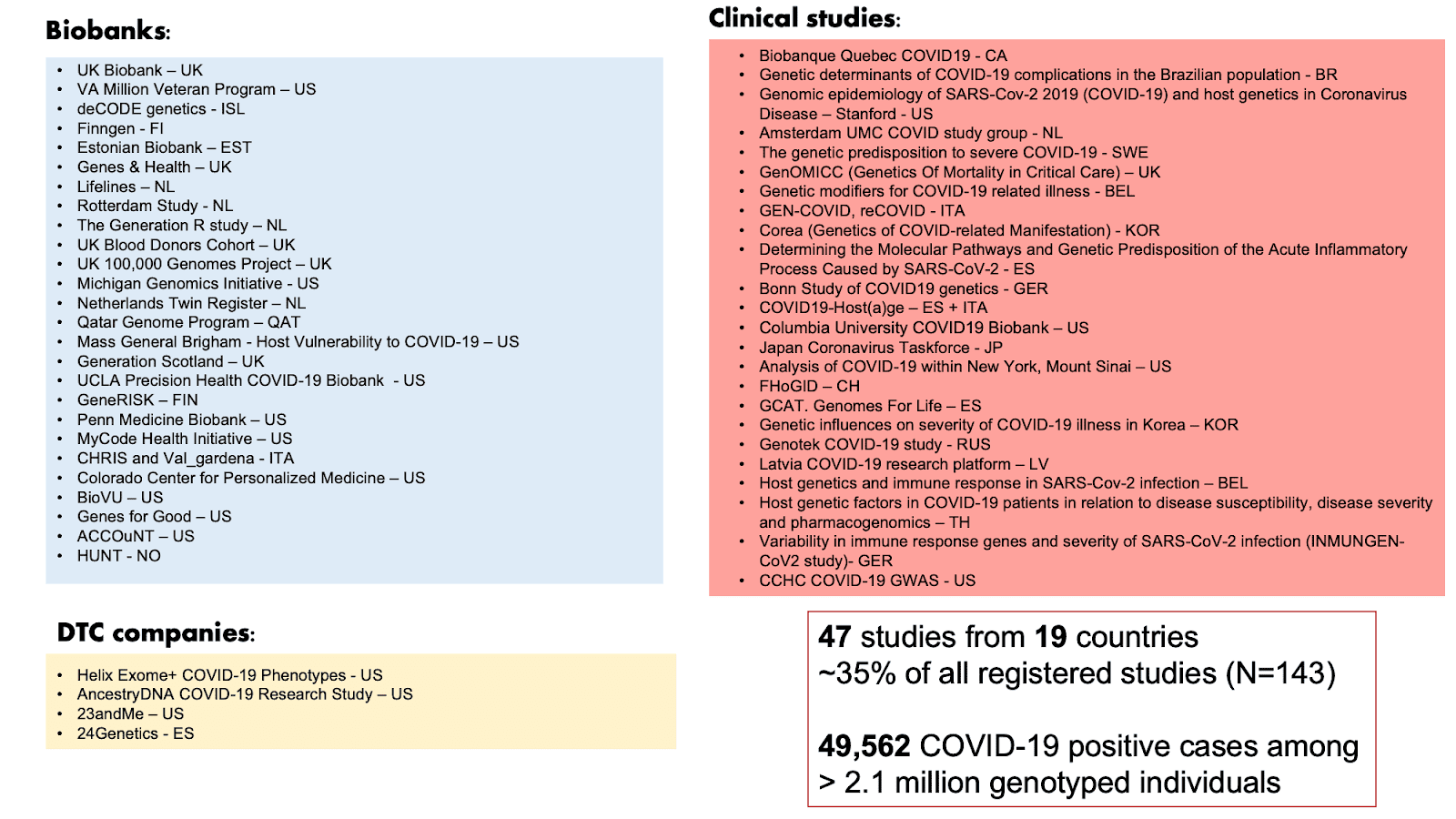
Figuur 1: Lijst van studies die deelnemen aan COVID-19 HGI in de release 5. Van de 47 deelnemende studies zijn er 19 die niet-Europese deelnemers hebben geïncludeerd. Gebaseerd op Andrea Ganna’s presentatie van 25 januari 2021.
Studie opzet
Net als in de vorige versie, analyseren we ook in deze versie drie verschillende uitkomstmaten (Figuur 2): A) Ernstig ziektebeloop van COVID-19 (beademing of overlijden aan COVID-19), B) Ziekenhuisopname (een maat voor de ‘ernst van de ziekte’) voor COVID-19, en C) Geïnfecteerd raken met het SARS-CoV-2 virus. Deze analyses hebben als doel om genetische factoren te onderzoeken die betrokken zijn bij de vatbaarheid voor SARS-CoV-2 en de ernst van ziekte door COVID-19 na infectie. De laatste analyse (analyse C) had als doel om genetische varianten te vinden die bijdragen aan het besmet raken met SARS-CoV-2. Analyse C bevatte alle cases, ongeacht óf en in welke mate zij hier klachten van hadden. De analyse uitkomsten, de case/control definities en studieomvang worden getoond in Figuur 2.

Figuur 2: Definitie van ‘cases’ (ziektegevallen) en controles voor de verschillende analyses in release 5. Opmerking: het SARS-CoV-2 virus veroorzaakt de COVID-19 infectie. Aangepast van Andrea Ganna’s presentatie op 25 januari 2021.
Regio's in het genoom die geassocieerd zijn met COVID-19 wijzen in de richting van het aangeboren immuunsysteem en longfunctie
Nadat we met hulp van onze collega wetenschappers de benodigde genetische data hadden verzameld (Figuur 1), voerden we een GWAS uit zoals beschreven in Figuur 2. Eerder, in release 4, belichtten we 7 nieuwe signalen (regio’s) in het genoom, die correleren met de vatbaarheid en ernst van COVID-19. Deze regio's wijzen in de richting van een rol voor het aangeboren immuunsysteem en longfunctie; dit past binnen de huidige klinische kennis dat bestaat over COVID-19 infecties. In deze nieuwe release 5 identificeerden we in totaal 15 significante regio's: 1 regio was alleen significant in de ‘ernstig ziektebeloop’ analyse (critically ill, analyse A); 11 regio's hadden een significante associatie met de ‘ziekenhuisopname’ een maat voor de ‘ernst van ziekte’ (hospitalized, analyse B); en 4 van de regio's zijn specifiek geassocieerd met het ‘geïnfecteerd raken met SARS-CoV-2’ (analyse C). In Figuur 3 zie je een Miami plot, dat is een gespiegelde versie van een Manhattan plot en een grafische weergave van de resultaten van analyse B en C. Het is een verwijzing naar de stad Miami, omdat de skyline van Miami op het water wordt weerspiegeld.
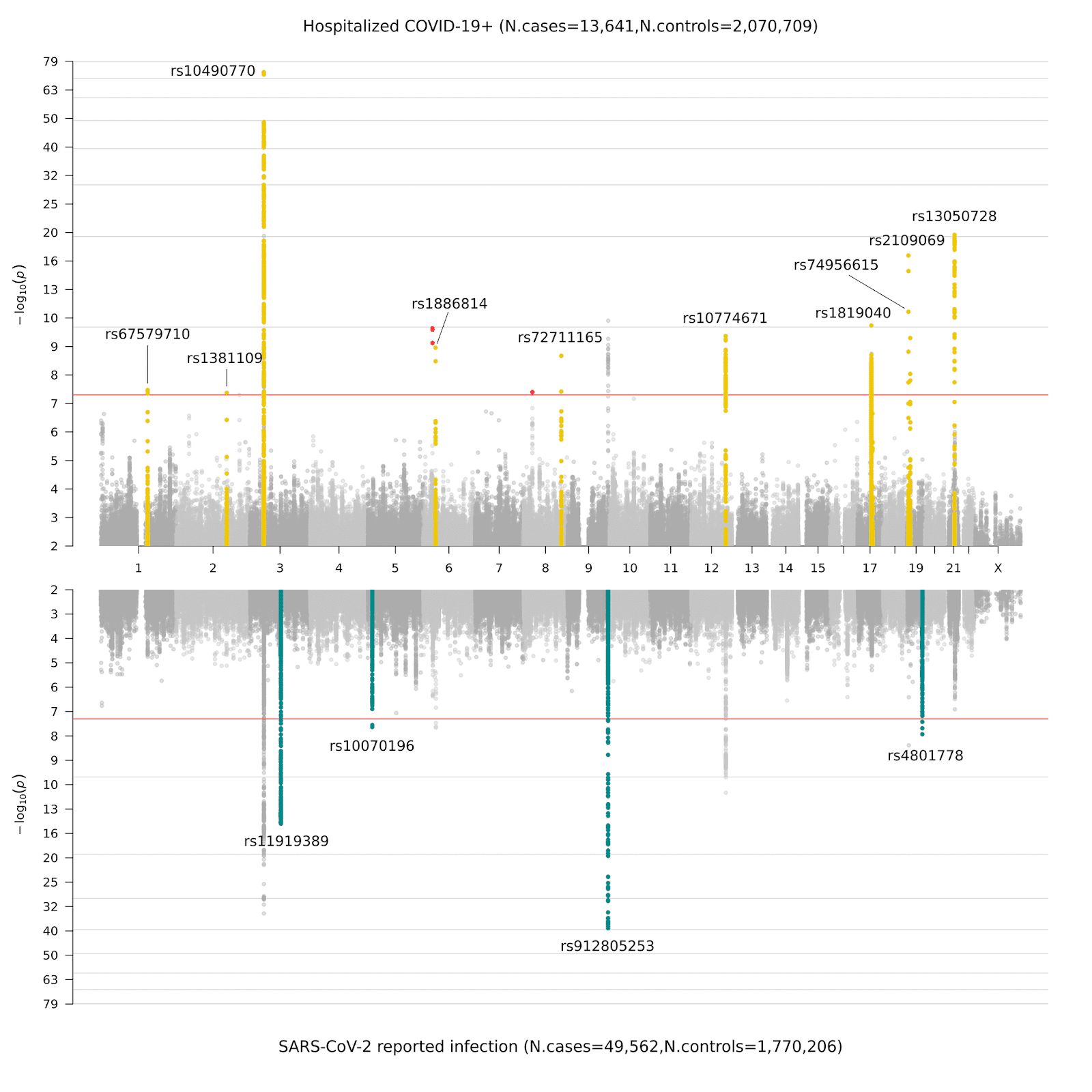
Figuur 3. Miami plot van de genoom-brede associatie resultaten voor COVID-19. Het bovenste deel laat de GWAS resultaten zien voor het ernstig ziektebeloop van COVID-19 (analysis B), en het onderste deel laat de resultaten zien van het risico op geïnfecteerd raken met SARS-CoV-2 (analysis C).
De rol van diversiteit in deze studie
In genetische studies is de diversiteit van bestudeerde deelnemers een belangrijk punt van aandacht (dit wordt hier verder toegelicht). Om deze reden hebben wij geprobeerd om de diversiteit van onze studie populatie te vergroten naarmate de omvang van de studie toenam (Figuur 4). Onze verbeterde dataverzameling heeft ertoe geleid dat nieuwe genetische factoren die betrokken zijn bij COVID-19 geïdentificeerd konden worden (onze vorige resultaten worden besproken in de blog posts van Release 3 en R4). Met het vinden van meerdere genetische factoren met onze analytische methoden, zijn we in staat om genetische varianten te identificeren die zich binnen of nabij een gen bevinden. Tot dusver zijn de meeste genen die we hebben geïdentificeerd betrokken bij mechanismen op celniveau, immuunregulatie en de werking van het hart. Het vinden van deze risico genen kan uiteindelijk bijdragen aan het vinden van nieuwe behandelingen.

Figuur 4. Overzicht van studies die hebben deelgenomen aan COVID-19-HGI en samenstelling van afkomst in de meta-analyse. In release 5 hebben 19 studies meegedaan van niet-Europese afkomst: 7 Afrikaans-Amerikaans, 5 gemengd Amerikaans, 4 Oost-Aziatisch, 2 Zuid-Aziatisch en 1 Arabisch. Diamant-vormige figuren geven de effectieve studie grootte weer van verschillende gebieden.
We vonden negen chromosoom regio’s die geassocieerd zijn met COVID-19. In analyse A (ernstig ziektebeloop) bevonden deze regio’s zich nabij twee genen: LZTFL1 op chromosoom 3 en TAC4 op chromosoom 17. Het LZTFL1 eiwit regelt transport van andere eiwitten naar de cilia membraan. Cilia zijn trilhaar structuren die uit het cellichaam steken. Deze trilharen bevinden zich in de luchtwegen, longen en vele andere organen. LZTFL1 vervult daarnaast ook een rol in de werking van het immuunsysteem. Het TAC4 eiwit reguleert de bloeddruk en de normale werking van het immuunsysteem.
Bij analyse B, in patiënten die opgenomen waren voor COVID-19, vonden we associaties nabij vier genen. Ten eerste vonden we een regio in THBS3 op chromosoom 1. Dit gen codeert voor het eiwit THBS3 dat tot expressie komt in het hart en verhoogd tot expressie komt bij hartziekten. Ten tweede vonden we een regio in SCN1A op chromosoom 2. Van het SCN1A gen is bekend dat mutaties in dit gen leiden tot epilepsie. Ten derde vonden we een regio in TMEM65 op chromosoom 8. Dit gen codeert voor het TMEM65 eiwit en speelt een rol in de aanleg, functie en geleidingssysteem van het hart. Daarnaast speelt dit eiwit een rol bij de energiehuishouding van de cel. De variant die in onze analyses werd gevonden verschilde interessant genoeg sterk in hoe vaak deze voorkomt in groepen van verschillende afkomst: de frequentie hiervan was 12% in mensen van Oost-Aziatische afkomst, en 1% in mensen van Europese afkomst. Ten slotte vonden we een regio in KANSL1 op chromosoom 17. In eerdere literatuur wordt gesuggereerd dat het eiwit dat door dit gen wordt aangemaakt, KANSL1, betrokken is bij processen in hersencellen.
Ten slotte, in analyse C waarbij we gerapporteerde SARS-CoV-2 infecties bestudeerden, vonden we drie nieuwe associaties in regio’s in de buurt van genen: ZBTB11 op chromosoom 3, DNAH5 op chromosoom 5, en PPP1R15A op chromosoom 19. Het ZBTB11 gen codeert voor het ZBTB11 eiwit, dat betrokken is bij de ontwikkeling van cellen in het immuunsysteem. Het verband met het DNAH5 gen op chromosoom 5 is interessant vanwege het feit dat dit gen ook betrokken is bij een ziektebeeld (primaire ciliaire dyskinesie) waarbij de trilharen van o.a. de luchtwegen niet goed werken. Dit leidt tot herhaaldelijke long ontstekingen, ontstekingen van de neus/oren/keel, bronchitis en onvruchtbaarheid. Tenslotte, de associatie met PPP1R15A op chromosoom 19: dit gen codeert voor het PPP1R15A eiwit dat verantwoordelijk is voor het remmen van de cel groei en het starten van celdood als reactie op DNA schade, remmende groeifactoren en afwijkende eiwit structuren.
Genen die het immuunsysteem beïnvloeden bleken een belangrijke rol te spelen bij COVID-19 in onze analyses. Daarnaast vonden we ook genen die betrokken zijn bij de werking van de longen en het hart, en bij processen in hersencellen. Hartziekten zijn in het verleden in verband gebracht als risicofactor voor COVID-19 en symptomen van het centraal zenuwstelsel zijn gerapporteerd als onderdeel van de symptomen bij COVID-19.
Een correlatie is niet hetzelfde als een oorzakelijk verband
Dit soort genetische studies identificeren genetische risicofactoren, maar dit levert nog geen bewijs van een oorzakelijke verband met het risico op COVID-19 of met de ernst van het ziekteverloop. Daarom hebben wij een methode toegepast die Mendeliaanse randomisatie (MR) wordt genoemd en die genetische informatie gebruikt om oorzakelijke verbanden af te leiden. MR is een methode die gebruik maakt van genetische varianten waarvan bekend is dat ze een bepaalde blootstelling beïnvloeden (bv. BMI) om het oorzakelijk verband van een blootstelling op ziekte-uitkomsten te onderzoeken. Meer weten over MR? We beschreven deze methode eerder in een blog, weliswaar gericht op een wetenschappelijke publiek. Kijkend naar de drie COVID-19 uitkomstmaten, ontdekten we oorzakelijke verbanden tussen de drie uitkomstmaten en 6 kenmerken (van de 38 geselecteerde kenmerken waarop we getest hebben, Figuur 4), die statistisch significant waren. Wij vonden bijvoorbeeld dat een genetisch voorspelde hogere body mass index (BMI, dat een indicatie geeft of er sprake is van ondergewicht of overgewicht) geassocieerd was met een hoger risico op SARS-CoV-2 infectie en COVID-19 ziekenhuisopname. Dit resultaat bevestigt bevindingen van epidemiologische studies, die vonden dat een verhoogd BMI in verband heeft met een verhoogd risico van ernstige COVID-19. Bovendien werd genetisch voorspeld roken geassocieerd met een verhoogd risico op COVID-19 ziekenhuisopname.
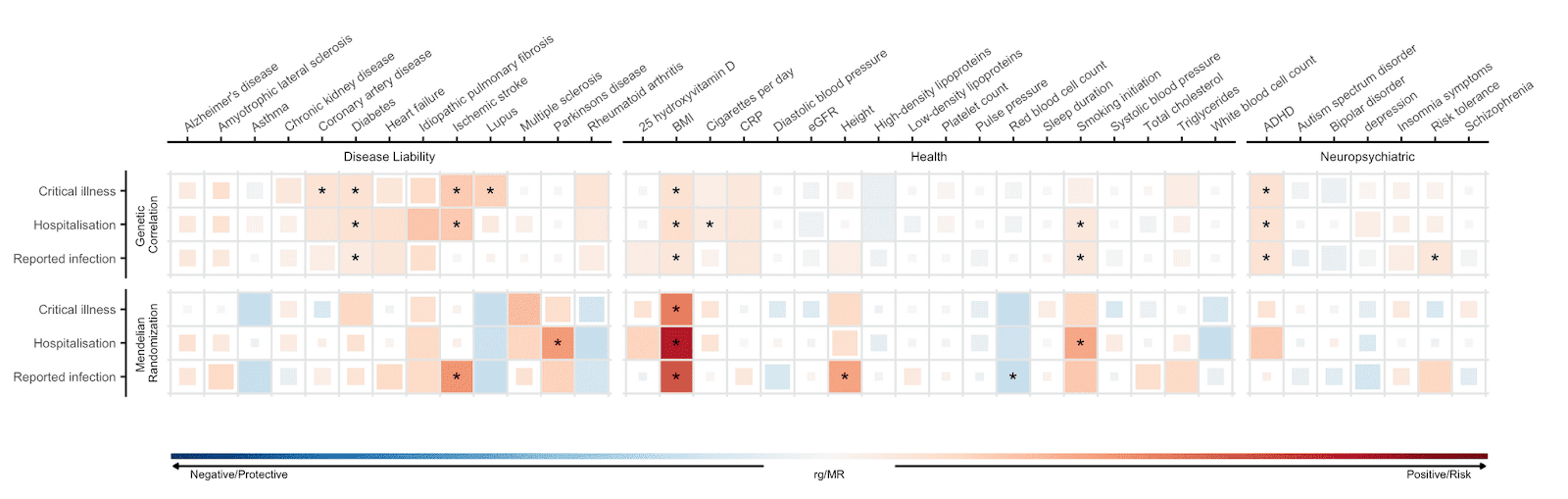
Figuur 5: Genetische correlaties en oorzakelijke verbanden tussen 38 kenmerken en de ernst van COVID-19 en de gerapporteerde SARS-CoV-2 infectie. De kenmerken staan op de X-as en de COVID-19 uitkomstmaten op de Y-as. Blauw staat voor een negatieve genetische correlatie en een beschermend oorzakelijk verband, ook wel ‘Mendeliaanse randomisatie (MR) causale schattingen’ genoemd, rood voor positieve genetische correlatie en risico-verhogende MR causale schattingen. Grotere vierkanten corresponderen met een sterkere significante correlatie. Statistisch significante causale schattingen zijn aangegeven met een asterisk.
Een wereldwijd samenwerkingsverband om de genetica van de COVID-19-gastheer beter te begrijpen
In de huidige wereldwijde crisis van de COVID-19 pandemie, tonen deze resultaten op basis van 47 individuele studies de kracht aan van de inspanning van honderden wetenschappers uit 19 verschillende landen. In totaal ontdekten wij 15 genomische regio's die sterk associëren met de gevoeligheid voor en de ernst van COVID-19. We pasten een statistische methode toe (Mendeliaanse randomisatie) om het oorzakelijk verband van deze regio's met het beloop en het risico op COVID-19 met 8 verschillende menselijke eigenschappen aan te tonen. Momenteel leggen we de laatste hand aan een artikel dat deze resultaten samenvat voor een wetenschappelijk tijdschrift. Naarmate we verder werken binnen COVID-19 HGI om een bijdrage te leveren aan de bestrijding van de wereldwijde COVID-19 pandemie, zullen wij steeds nieuwe genetische resultaten presenteren. Door samen te werken en te delen, kunnen we de betrouwbare resultaten verkrijgen, die nodig zijn om de biologie en de klinische verschijnselen van COVID-19 beter te begrijpen.
Dankbetuiging
Dank aan Andrea Ganna voor de bedachtzame feedback en het verhelderende commentaar.