This is a translation in Portuguese. You can also read the original English version.
portugese-3rd-blog
April 28, 2021
Resultados de COVID-19 HGI para congelamento de dados 5 (janeiro de 2021)
02 de março de 2021
Escrito por Minttu Marttila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford e Kumar Veerapen em nome do COVID-19 HGI
Nota: A COVID-19 Host Genetics Initiative (HGI) representa um consórcio de mais de 2.000 cientistas de mais de 54 países trabalhando em colaboração para compartilhar dados, ideias, recrutar pacientes e divulgar nossas descobertas. Para obter uma cartilha sobre o projeto do nosso estudo, leia nossa postagem inaugural do blog (inaugural blog post). Nossa pesquisa é iterativa e resumimos nossos novos resultados por meio de postagens de blog (blog posts) e na seção de resultados (results) de nosso site. Finalmente, se algum vocabulário aqui for desconhecido, envie-nos um e-mail para hgi-faq@icda.bio— ficaremos felizes em atualizar as informações aqui para fornecer mais clareza. Nas próximas semanas, serão disponibilizadas informações adicionais explicando conceitos ou terminologia. Nesse ínterim, dê uma olhada neste (this) recurso para revisar os fundamentos da genética
O artigo científico desta versão está agora no medRXiv.
O congelamento de dados mais recente (versão 5) aumenta o tamanho da amostra e a robustez das descobertas genéticas
O COVID-19 HGI mostrou iterativamente sinais genéticos robustos em nossas versões anteriores e representa o maior estudo de associação do genoma (genome-wide association study) (GWAS) da história: tanto em termos de participantes do estudo (> 2 milhões de indivíduos) e número de colaboradores (> 2.000 cientistas ) Aqui, descrevemos os resultados de nosso último congelamento de dados, versão 5. Em nosso congelamento de dados anterior (versão 4), relatamos a identificação de variantes genéticas humanas associadas a COVID-19 grave (veja nossas postagens no blog para não cientistas no lançamento de dados 3 (release 3) e versão 4 (release 4)). Identificamos essas variantes por meio de um GWAS em mais de 30.000 pacientes COVID-19 (ou seja, casos) e 1,47 milhões de pacientes não COVID-19 (ou seja, controles). No congelamento de dados 5, aumentamos ainda mais o tamanho da amostra para quase 50.000 casos COVID-19 e mais de 2 milhões de controles, combinando dados de 47 estudos em 19 países (Figura 1). Ao aumentar o tamanho da amostra, também estamos melhorando a confiança de nossas descobertas. Nesse congelamento de dados, também tentamos melhorar a diversidade das populações. O estudo da genética em populações de diversas linhagens genéticas nos ajuda a entender melhor as variantes genéticas que afetam a gravidade do COVID-19 e seu impacto em todo o mundo. Dos 47 estudos contribuintes, 19 incluíram populações não europeias.

Figura 1: Lista de contribuintes COVID-19 HGI para liberação de congelamento de dados 5 Dos 47 estudos colaboradores, 19 incluíram populações não europeias. Adaptado da apresentação de Andrea Ganna’s presentation em 25 de janeiro de 2021.
Estrutura do Estudo
Como nos congelamentos de dados anteriores, continuamos a examinar três desfechos (Figura 2): A) Estar gravemente doente com COVID-19 (necessidade de suporte respiratório ou morrer por COVID-19), B) Ser hospitalizado por COVID-19 e C ) Estar infectado com SARS-CoV-2. Essas análises visam capturar características genéticas associadas à suscetibilidade e gravidade ao SARS-CoV-2 e COVID-19. A última análise (Análise C) teve como objetivo detectar variantes genéticas que contribuem para a infecção relatada de SARS-CoV-2. Esta análise incluiu todos os casos, independentemente da presença ou gravidade dos sintomas. Os resultados da análise e suas definições de caso e controle e tamanhos de amostra são mostrados na Figura 2.
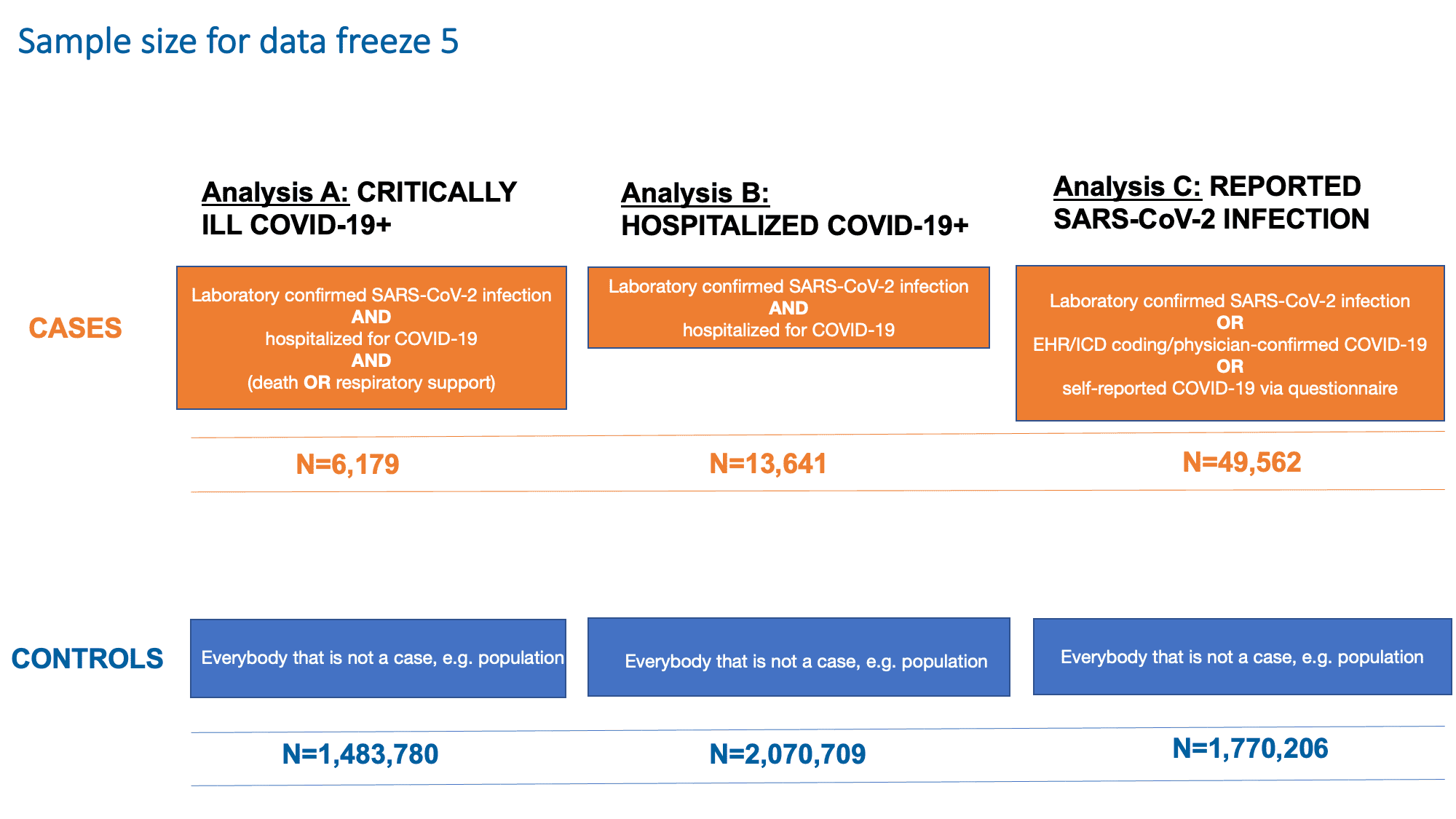
Figura 2: Definição de casos e controles para cada uma das análises no congelamento de dados 5. Observe que o SARS-CoV-2 é o vírus que causa a infecção COVID-19. Adaptado de Andrea Ganna’s presentation em 25 de janeiro de 2021.
Regiões do genoma associadas a COVID-19 apontam para imunidade inata e disfunção pulmonary
Ao coletar os dados genéticos produzidos por nossos colaboradores, realizamos GWAS de acordo com as definições na Figura 2. Anteriormente, no congelamento de dados 4 (data freeze 4), destacamos novos sinais genéticos na suscetibilidade e gravidade de COVID-19 em 7 regiões cromossômicas. Essas regiões apontam para uma etiologia na imunidade inata e disfunção pulmonar: em linha com a compreensão clínica líder das infecções por COVID-19. No congelamento de dados 5, identificamos 15 regiões significativas em todo o genoma em todos os cromossomos: 1 região cromossômica teve significância em todo o genoma apenas na análise criticamente doente (análise A); 11 regiões cromossômicas têm maior efeito na análise de gravidade do que na análise de infecção relatada (análise B); e 4 dessas regiões cromossômicas são específicas para infecção relatada por SARS-CoV-2 (análise C). Na Figura 3, apresentamos uma representação gráfica desses resultados como um gráfico de Miami (uma versão em painel de um Manhattan plot. Nomeado em homenagem a Miami porque a silhueta dos prédios de Miami se refletem na água).
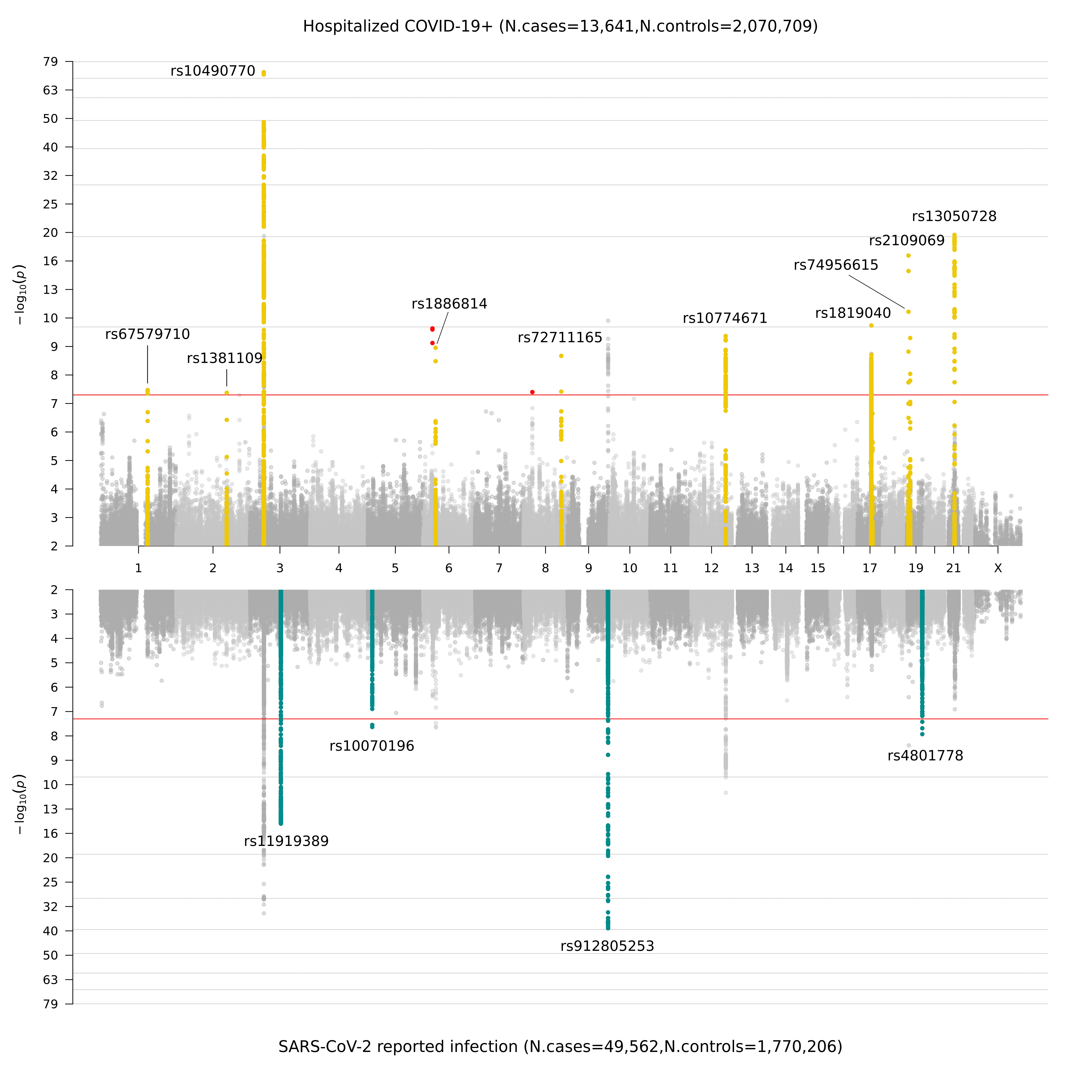
Figura 3. Gráfico de Miami dos resultados de associação do genoma para COVID-19. O painel superior mostra os resultados do estudo de associação do genoma completo de pacientes COVID-19 hospitalizado e controles (análise B), e o painel inferior mostra os resultados da infecção por SARS-CoV-2 relatada e controles (análise C).
A diversidade de amostras
Entendemos que, com muitos estudos genéticos, a diversidade na coleta de amostras é uma grande preocupação (elaborada aqui) (elaborated here). Como tal, nosso objetivo era melhorar a diversidade em nossa coleção de amostras à medida que nosso estudo crescia (Figura 4). Nosso esforço de coleta de amostras aprimorado nos levou a identificar novos fatores genéticos associados ao COVID-19 (nossos resultados anteriores são discutidos em postagens no blog nas versões 3 e R4) (blog posts on Release 3 and R4). Com a identificação concomitante de fatores de risco genéticos usando nossos métodos analíticos, somos capazes de observar variantes genéticas nos genes ou próximas a eles. Até agora, a maioria dos genes que identificamos apontam para um risco aumentado nos mecanismos celulares, regulação imunológica e função cardíaca. A identificação desses fatores de risco pode levar a tratamentos direcionados aos genes identificados.
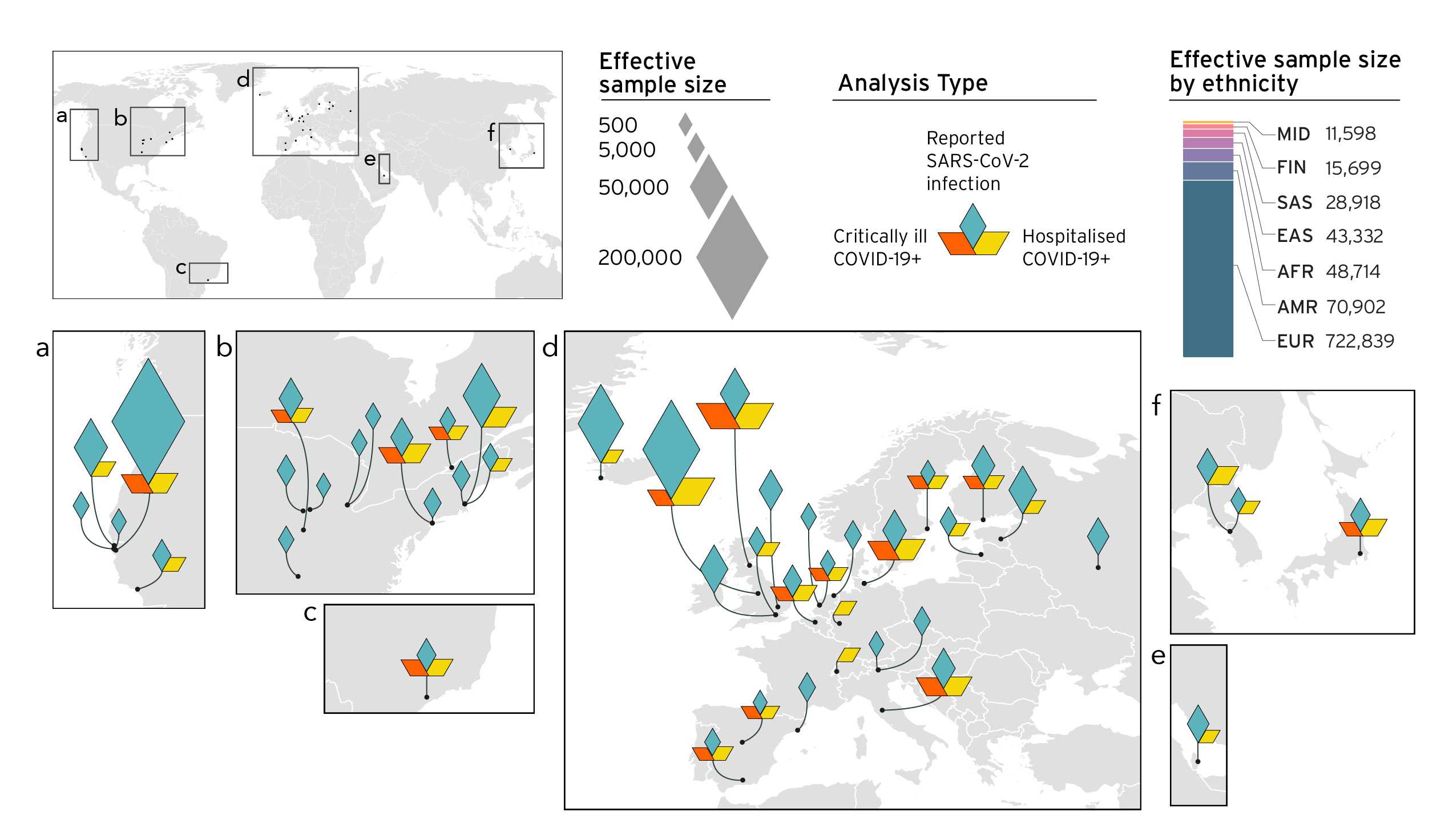
Figura 4. Visão geral dos estudos que contribuem para a iniciativa COVID-19 para composição genética do hospedeiro pelas principais ancestralidades que entraram na meta-análise. No congelamento de dados 5, 19 estudos contribuíram com populações não europeias: 7 afro-americanos, 5 americanos mistos, 4 do leste asiáticos, 2 sul asiáticos e 1 árabe. Os diamantes mostram o tamanho efetivo da amostra (tamanho da amostra que encontrará efeito estatisticamente significativo em eventos científicos) recebida de diferentes localizações geográficas.
Encontramos 9 novas regiões cromossômicas associadas ao COVID-19. Na análise A, para doença crítica, isso inclui regiões cromossômicas próximas a dois genes: LZTFL1 no cromossomo 3 e TAC4 no cromossomo 17. A proteína LZTFL1 regula o tráfego de proteínas para a membrana ciliar (raiz: cílios). Os cílios são estruturas semelhantes a cabelos que se estendem do corpo celular. Eles são encontrados nas vias respiratórias, pulmões e muitos outros órgãos. LZTFL1 também participa das respostas imunológicas. A proteína TAC4 funciona para regular a pressão arterial e o sistema imunológico.
Para a Análise B, em pacientes hospitalizados com COVID-19, encontramos associações em variantes próximas a 4 genes. Em primeiro lugar, identificamos uma região cromossômica em THBS3 no cromossomo 1. Esse gene codifica a proteína THBS3 que é expressa no coração e regulada positivamente durante doenças cardíacas. Em segundo lugar, identificamos uma região cromossômica em SCN1A no cromossomo 2. Foi demonstrado que variações no gene SCN1A causam epilepsia e convulsões. Em terceiro lugar, identificamos uma região cromossômica no TMEM65 no cromossomo 8. Este gene codifica a proteína TMEM65, que tem um papel no desenvolvimento cardíaco, regulação da condução e função cardíaca. Também pode desempenhar um papel no metabolismo da energia celular. Notavelmente, a variante identificada em nossa análise para TMEM65 tem frequência de 12% no Leste Asiático e 1% na população europeia. As frequências alélicas descrevem a quantidade de variação em um determinado gene ou região genômica. Finalmente, identificamos uma região cromossômica em KANSL1 no cromossomo 17. Foi sugerido que a proteína codificada por este gene, KANSL1, tem um papel em processos neuronais.
Finalmente, na análise C, para infecções relatadas de SARS-CoV-2, 3 novas associações foram encontradas nas regiões próximas aos genes, ZBTB11 no cromossomo 3, DNAH5 no cromossomo 5 e PPP1R15A no cromossomo 19. Em primeiro lugar, identificamos uma região próxima ao gene ZBTB11 no cromossomo 3. Esse gene codifica a proteína ZBTB11, que regula o desenvolvimento de células imunes. Em segundo lugar, identificamos uma região cromossômica no DNAH5 no cromossomo 5. Foi demonstrado que variações genéticas no DNAH5 causam discinesia ciliar primária, movimento defeituoso dos cílios, levando a infecções pulmonares recorrentes, sintomas de ouvido / nariz / garganta, bronquite e infertilidade. Finalmente, identificamos uma região cromossômica próxima a PPP1R15A no cromossomo 19. Esse gene codifica a proteína PPP1R15A que demonstrou mediar a interrupção do crescimento e a morte celular em resposta a danos no DNA, sinais negativos de crescimento e estrutura incorreta da proteína.
Os genes que afetam o sistema imunológico desempenham um papel importante no COVID-19 em nossas análises. Os genes envolvidos na função pulmonar e cardíaca e nos processos neuronais também fazem parte de nossas descobertas. As doenças cardíacas foram relatadas anteriormente como um fator de suscetibilidade ao COVID-19 e os sintomas neuronais relatados como parte da doença COVID-19.
Correlação não significa causalidade
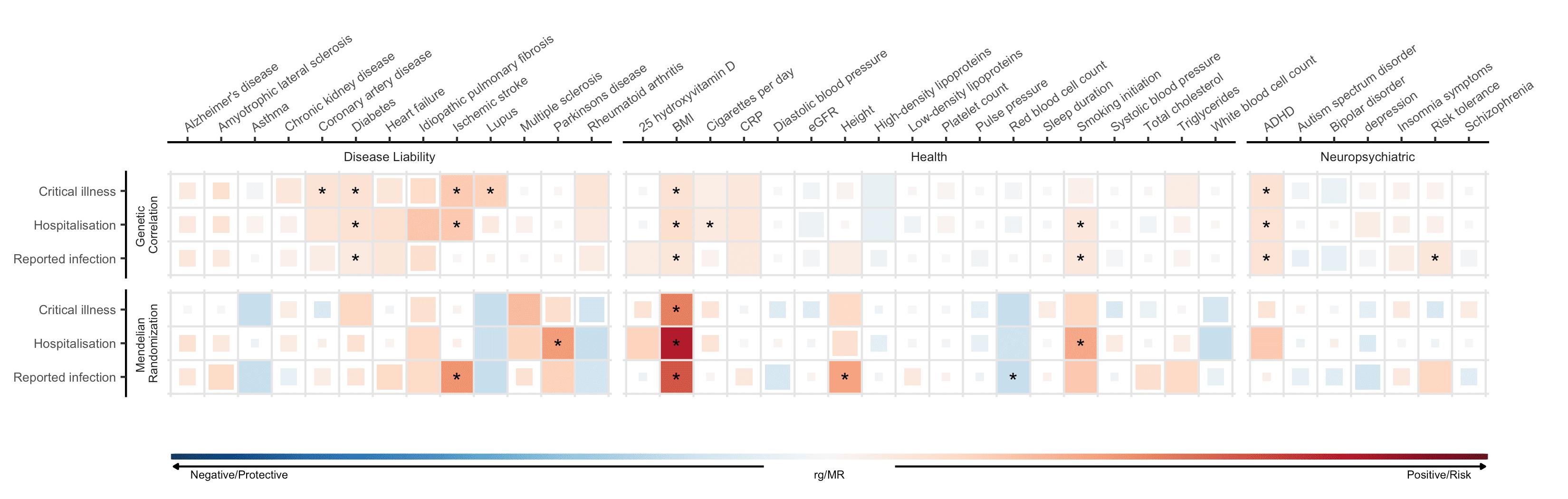
Os fatores de risco identificados em estudos de associação podem não apontar para uma base causal da suscetibilidade ou gravidade do COVID-19. Como tal, empregamos um método chamado randomização Mendeliana (RM) Mendelian randomization (MR), que usa informações genômicas para inferir associações causais. MR é um método que usa variantes genéticas conhecidas por influenciar uma determinada exposição (por exemplo, IMC) para examinar o efeito causal de uma exposição nos resultados da doença. Para uma análise mais detalhada, descrevemos o MR em uma postagem recente (blog post) (voltada para o público científico). Entre os três fenótipos COVID-19, identificamos associações causais estatisticamente significativas entre os três resultados COVID-19 e 6 características (de 38 características selecionadas que testamos, Figura 4). Descobrimos que o maior índice de massa corporal (IMC) previsto geneticamente foi associado a um maior risco de infecção por SARS-CoV-2 e hospitalização por COVID-19. Esse resultado corrobora os achados de estudos observacionais, que observaram um risco aumentado de desfechos COVID-19 graves associados ao IMC aumentado. Além disso, o tabagismo previsto geneticamente foi associado a um risco aumentado de hospitalização por COVID-19.
{kind=link}
Figura 5: Correlações genéticas e estimativas causais de randomização mendeliana entre 38 características e gravidade de COVID-19 e infecção relatada por SARS-CoV-2. As características são listadas no eixo X e fenótipos COVID-19 no eixo Y. Azul representa correlação genética negativa e estimativas causais de randomização mendeliana protetora (MR) e vermelho representa correlação genética positiva e estimativas causais de risco MR. Quadrados maiores correspondem a correlação mais significativa. As estimativas causais que ultrapassam um limite de significância estatística são marcadas com um asterisco.
Um esforço colaborativo global para entender a genética do hospedeiro na COVID-19
Na atual crise global da pandemia COVID-19, esses resultados demonstram o poder de um esforço global de 47 colaboradores de diversas partes do mundo. No total, identificamos 15 regiões genômicas associadas à suscetibilidade e gravidade de COVID-19. Para interrogar ainda mais a causalidade dessas regiões, utilizamos inferência estatística (ou seja, randomização mendeliana) para identificar 6 características com causalidade estatisticamente significativa com esses sinais COVID-19 GWAS. Atualmente, estamos finalizando nossos resultados em um artigo científico. À medida que continuamos a enfrentar a pandemia global COVID-19, o COVID-19 HGI produzirá resultados genéticos iterativamente. Trabalhando juntos, podemos gerar achados robustos necessários para melhor compreender os fatores biológicos e a apresentação clínica do COVID-19.
Agradecimentos
Obrigado a Andrea Ganna, PhD, por seus comentários e feedbacks.
Translated by J.E. Krieger and A.C. Pereira.